Modeling Quantum Noise for Accurate Chemistry Simulations: A Guide to Depolarizing and Amplitude Damping Channels in Drug Discovery
This article provides a comprehensive guide for researchers and drug development professionals on the critical role of quantum noise models, specifically depolarizing and amplitude damping channels, in computational chemistry simulations...
Modeling Quantum Noise for Accurate Chemistry Simulations: A Guide to Depolarizing and Amplitude Damping Channels in Drug Discovery
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on the critical role of quantum noise models, specifically depolarizing and amplitude damping channels, in computational chemistry simulations on Noisy Intermediate-Scale Quantum (NISQ) devices. We explore the fundamental physics behind these noise types, their distinct impacts on quantum algorithms like VQE, and methodological approaches for their integration into chemical simulation workflows. The content further covers advanced error mitigation strategies, validation techniques for assessing model accuracy, and comparative analyses of noise resilience across different algorithmic approaches. By synthesizing the latest research and practical implementation strategies, this work aims to equip scientists with the knowledge needed to develop more reliable and predictive quantum chemistry simulations for biomedical applications.
Understanding Quantum Noise: The Fundamental Challenge in Chemical Simulation on NISQ Devices
The Noisy Intermediate-Scale Quantum (NISQ) era represents the current technological frontier in quantum computing, characterized by processors containing 50 to 1,000 physical qubits that operate without full error correction [1]. For quantum chemistry applications, including molecular energy estimation and drug discovery, these devices present both unprecedented opportunities and fundamental limitations. This technical guide analyzes the core constraints of NISQ hardware through the lens of quantum noise models relevant to chemistry simulations, detailing experimental methodologies for overcoming these limitations while providing quantitative resource assessments and practical toolkits for researchers in pharmaceutical and materials science domains.
Fundamental NISQ Hardware Limitations
NISQ devices are defined by strict physical constraints that directly impact their utility for quantum chemistry applications. These limitations emerge from current technological boundaries in qubit fabrication, control, and maintenance of quantum coherence.
Physical Resource Constraints
Quantum computers in the NISQ era operate with severe resource restrictions that bound the complexity of executable algorithms [2]. The table below summarizes the key hardware limitations across major qubit platforms:
Table 1: NISQ Hardware Performance Metrics Across Platforms
| Platform | Qubit Count Range | 2-Qubit Gate Fidelity (%) | Coherence Times (Tâ‚/Tâ‚‚) | Gate Time |
|---|---|---|---|---|
| Superconducting (e.g., IBM, Google) | 27-1000+ | 98.6-99.7 [3] | 10-100 μs [3] | 20-100 ns [3] |
| Trapped Ions (e.g., IonQ, Quantinuum) | 11-50 | 99.8-99.9 [3] | 1-10 seconds [3] | 50-200 μs [3] |
| Neutral Atoms (e.g., Pasqal) | ~100 | 97-99 [3] | 0.1-1 seconds [3] | ~1 ms [3] |
The total number of gates executable before decoherence dominates is determined by N·d·ε ≪ 1, where N is the qubit count, d is the circuit depth, and ε is the two-qubit error rate [3]. This relation fundamentally constrains the algorithmic complexity achievable on NISQ devices.
Quantum Noise Models and Their Impact
For quantum chemistry simulations, understanding specific noise models is essential for developing effective error mitigation strategies. The most prevalent models include:
Unital Noise Models (Depolarizing Noise)
Depolarizing noise represents a symmetric, unital noise model that randomly replaces the current state with the maximally mixed state with probability p. For a single qubit, this channel can be represented as: Λâ‚(Ï) = (1-p)Ï + p(I/2) [4]
This model is strictly contractive and drives any quantum state toward the maximally mixed state [4]. Under such noise, the relative entropy between the state Ï(t) and the maximally mixed state diminishes as D(Ï(t)∥σ₀) ≤ nμᵗ, where σ₀ = I/2â¿ is the n-qubit maximally mixed state and μ < 1 is the contractive rate [4]. For quantum chemistry applications, this presents particular challenges for maintaining coherent superposition states essential for molecular orbital simulations.
Non-Unital Noise Models (Amplitude Damping)
Amplitude damping represents non-unital noise that models energy dissipation, a physically relevant model for molecular systems. Unlike unital noise, amplitude damping has a unique fixed point and does not necessarily drive all states toward the maximally mixed state [5]. Recent research has identified that while unital noise always induces Noise-Induced Barren Plateaus (NIBPs), Hilbert-Schmidt contractive non-unital noise (including amplitude damping) does not necessarily lead to barren plateaus, suggesting some noise types may be less detrimental to variational quantum algorithms [5].
Diagram Title: NISQ Noise Model Effects on Quantum States
Algorithmic Limitations for Chemistry Applications
Theoretical Performance Boundaries
Recent complexity-theoretic results establish fundamental limitations for NISQ devices in quantum chemistry applications. For strictly contractive unital noise, quantum devices become statistically indistinguishable from random circuits when depths exceed Ω(log(n)) [4]. Even with classical processing, devices with super-logarithmic circuit depths fail to deliver quantum advantage for polynomial-time algorithms [4].
Spatial connectivity constraints further limit performance. For one-dimensional noisy qubit circuits, super-polynomial quantum advantages are ruled out in all-depth regimes, with entanglement generation capped at O(log(n)) for 1D circuits and O(√nlog(n)) for 2D circuits [4]. These bounds directly impact the simulation of complex molecular systems requiring high entanglement.
Practical Algorithmic Constraints
Variational Quantum Eigensolver (VQE), the leading NISQ algorithm for quantum chemistry, faces multiple practical constraints:
- Measurement Overhead: Resource estimates scale as S ∼ O(Nâ´/ϵ²) shots per iteration to achieve chemical accuracy [3]
- Gate Fidelity Requirements: Achieving chemical accuracy requires gate fidelities of ε < 10â»â´ to 10â»â¶ [3]
- Noise-Induced Barren Plateaus: Both unital and certain non-unital noise models cause exponentially small gradients in variational algorithms [5]
- Circuit Depth Limits: Current hardware constraints cap circuit depths at O(10²-10³) gates [3]
Table 2: Resource Requirements for Chemical Accuracy in Molecular Simulations
| Molecule | Qubits Required | Pauli Terms | Circuit Depth | Estimated Shots |
|---|---|---|---|---|
| BODIPY-4 (8e8o) | 8 | 361 [6] | ~10² | 10âµ-10ⶠ|
| BODIPY-4 (14e14o) | 28 | 55,323 [6] | ~10³ | 10â·-10⸠|
| FeMoco (Nitrogenase) | ~1,000,000 (estimated) [7] | ~10¹² (estimated) | >10ⷠ| >10¹ⵠ|
Experimental Methodologies and Protocols
High-Precision Measurement Techniques
Achieving chemical precision (1.6×10â»Â³ Hartree) on NISQ devices requires specialized measurement protocols. Recent experimental demonstrations on IBM Eagle processors have reduced measurement errors from 1-5% to 0.16% through integrated techniques [6]:
Protocol 1: Locally Biased Random Measurements
This technique reduces shot overhead by preferentially selecting measurement settings that have greater impact on energy estimation. The protocol maintains the informationally complete nature of measurements while optimizing resource allocation:
- Initialization: Prepare Hartree-Fock state on quantum processor
- Hamiltonian Segmentation: Decompose molecular Hamiltonian into Pauli terms
- Setting Selection: Apply biased selection toward high-weight Pauli terms
- Execution: Implement selected measurement settings on hardware
- Iteration: Update bias based on preliminary results
Protocol 2: Quantum Detector Tomography with Repeated Settings
This approach addresses circuit overhead and readout errors through parallel quantum detector tomography:
- Circuit Design: Implement informationally complete measurement circuits
- Parallel Tomography: Execute quantum detector tomography circuits alongside main computation
- Noise Characterization: Construct noisy measurement effects matrix P(m|s)
- Inversion: Apply Pâ»Â¹ to raw measurements to mitigate readout errors
- Validation: Verify mitigation efficacy through consistency checks
Protocol 3: Blended Scheduling for Time-Dependent Noise
Temporal noise variations pose significant barriers to high-precision measurements. Blended scheduling intersperses different circuit types to mitigate time-dependent noise effects:
- Circuit Grouping: Organize circuits by type (QDT, Hamiltonian terms)
- Temporal Interleaving: Execute circuits from different groups in sequence
- Noise Tracking: Monitor temporal noise patterns via calibration circuits
- Post-Processing: Apply temporal noise correction models
Diagram Title: High-Precision Measurement Workflow
Solvent-Ready Algorithm Implementation
Recent advances enable simulation of solvated molecules, critical for biologically relevant chemistry. The SQD-IEF-PCM (Sample-based Quantum Diagonalization with Integral Equation Formalism Polarizable Continuum Model) protocol represents a significant step toward practical quantum chemistry [8]:
- Wavefunction Sampling: Generate electronic configurations from molecular wavefunction using quantum hardware
- Noise Correction: Apply self-consistent process (S-CORE) to restore physical properties (electron number, spin)
- Subspace Construction: Build manageable subspace from corrected configurations for classical processing
- Solvent Incorporation: Add solvent effect as perturbation to molecular Hamiltonian using IEF-PCM
- Self-Consistent Iteration: Update molecular wavefunction until solvent and solute reach mutual consistency
Experimental implementation on IBM quantum computers with 27 to 52 qubits has demonstrated solvation free energies matching classical benchmarks within 0.2 kcal/mol for methanol [8].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Experimental Components for NISQ Quantum Chemistry
| Component | Function | Implementation Example |
|---|---|---|
| Informationally Complete (IC) Measurements | Enables estimation of multiple observables from same measurement data [6] | Locally biased random measurements for reduced shot overhead |
| Quantum Detector Tomography (QDT) | Mitigates readout errors by characterizing noisy measurement effects [6] | Parallel QDT execution alongside main quantum circuits |
| Error Mitigation Techniques | Improves result accuracy without quantum error correction [1] | Zero-noise extrapolation, probabilistic error cancellation, symmetry verification |
| Hybrid Quantum-Classical Algorithms | Leverages classical resources to reduce quantum circuit demands [9] | VQE for ground state energy estimation, QAOA for optimization |
| Implicit Solvent Models | Incorporates solvent effects without explicit solvent molecules [8] | IEF-PCM for solvation energy calculation in aqueous solutions |
| Dynamic Compilation | Adapts circuits to hardware constraints and noise profiles [3] | Calibration-aware qubit placement and routing |
| Ki16425 | Ki16425, CAS:355025-24-0, MF:C23H23ClN2O5S, MW:475.0 g/mol | Chemical Reagent |
| L-167307 | L-167307, CAS:188352-45-6, MF:C22H17FN2OS, MW:376.4 g/mol | Chemical Reagent |
Future Perspectives: Beyond the NISQ Era
The transition from NISQ to Fault-Tolerant Application-Scale Quantum (FASQ) computing requires overcoming multiple technological hurdles. Current estimates suggest that a modest 1,000 logical-qubit processor would require approximately one million physical qubits with current error rates [10]. While error mitigation techniques provide a temporary bridge, their sampling overhead grows exponentially with circuit size [10].
Promising developments include new qubit designs such as fluxonium and cat qubits that may reduce error correction overhead, and algorithmic advances that identify "proof pockets" - specific subproblems where quantum methods demonstrate clear advantages [10]. For quantum chemistry, the first practical applications are likely to emerge in specialized simulation domains before expanding to broader commercial applications in pharmaceutical and materials science [10].
The scientific toolkit presented herein provides researchers with practical methodologies for extracting meaningful chemical insights from current NISQ devices while the field progresses toward fault-tolerant quantum computation capable of addressing industrially relevant molecular systems.
This technical guide provides a comprehensive examination of depolarizing noise, a critical model for describing non-coherent errors in quantum information processing. We present the complete mathematical formulation of the depolarizing channel using Kraus operators and detail its impact on quantum systems. Framed within quantum chemistry simulations, this review explores how depolarizing noise and other error models affect the accuracy of quantum computations for chemical systems, contrasting its universally detrimental effects with the context-dependent impacts of amplitude damping noise. We further synthesize current experimental protocols for noise characterization and mitigation, providing researchers with essential tools for evaluating and countering noise in near-term quantum devices.
Quantum computation holds revolutionary potential for simulating molecular systems and chemical reactions, a task that remains intractable for classical computers as system size increases [11] [12]. However, current quantum processors operate in the noisy intermediate-scale quantum (NISQ) era, where quantum information is highly susceptible to corruption from environmental interactions and imperfect control [13] [14]. These disruptive processes are formally described using quantum noise models—mathematical representations of how quantum states evolve when coupled to an external environment.
The most prevalent noise models include:
- Depolarizing Noise: A non-coherent, unital process that randomly applies Pauli operators to a qubit with equal probability.
- Amplitude Damping: A non-unital process modeling energy dissipation from an excited state to a ground state.
- Phase Damping: A coherent dephasing process that destroys phase information without energy loss.
Understanding these models, particularly their mathematical structure through Kraus operators and their physical implications for quantum algorithms, is prerequisite for developing effective error suppression and correction techniques in quantum computational chemistry.
Mathematical Formulation of the Depolarizing Channel
Kraus Operator Formalism
Quantum noise processes are formally described by completely positive trace-preserving (CPTP) maps, which guarantee the physical validity of the transformed quantum state. The most general representation of a CPTP map is the Kraus operator sum representation:
$$ \mathcal{E}(\rho) = \sum{k} A{k} \rho A_{k}^{\dagger} $$
where $\rho$ is the density matrix of the quantum state, and the Kraus operators $A{k}$ satisfy the completeness relation $\sum{k} A{k}^{\dagger} A{k} = I$ to ensure trace preservation [15].
Single-Qubit Depolarizing Channel
The single-quit depolarizing channel represents a process where the qubit remains unchanged with probability $1-p$, or undergoes randomization via Pauli $X$, $Y$, or $Z$ errors with equal probability $p/3$. This channel is described by four Kraus operators:
$$ \begin{aligned} A0 &= \sqrt{1-p} \, I \ A1 &= \sqrt{\frac{p}{3}} \, X \ A2 &= \sqrt{\frac{p}{3}} \, Y \ A3 &= \sqrt{\frac{p}{3}} \, Z \end{aligned} $$
where $I, X, Y, Z$ are the Pauli matrices. The action of the depolarizing channel on a quantum state $\rho$ is therefore:
$$ \mathcal{E}_{\text{depol}}(\rho) = (1-p)\rho + \frac{p}{3}(X\rho X + Y\rho Y + Z\rho Z) $$
This formulation reveals the depolarizing channel as a Pauli channel with equal weights for all non-identity Pauli operations. For a single qubit, the depolarizing channel can be alternatively expressed as:
$$ \mathcal{E}_{\text{depol}}(\rho) = (1-p)\rho + p \frac{I}{2} $$
which clearly shows the state being replaced with the maximally mixed state $I/2$ with probability $p$.
Multi-Qubit and Correlated Depolarizing Noise
For multi-qubit systems, the depolarizing channel can be generalized to include both local depolarizing noise (affecting qubits independently) and correlated depolarizing noise (simultaneously affecting multiple qubits). The cluster expansion approach provides a systematic framework for constructing approximate noise channels by incorporating noise components with increasing degrees of qubit-qubit correlation [16].
A $k$-th order approximate channel incorporates correlations up to degree $k$, with the full noise model requiring resources growing exponentially with the number of qubits. This approach enables honest noise modeling—where actual errors are not underestimated—which is crucial for predicting the performance of quantum error correction codes.
Table 1: Key Properties of Common Single-Qubit Noise Channels
| Noise Channel | Kraus Operators | Type | Qubit Effect | Invertible | ||
|---|---|---|---|---|---|---|
| Depolarizing | $A0 = \sqrt{1-p}I$, $A1 = \sqrt{p/3}X$, $A2 = \sqrt{p/3}Y$, $A3 = \sqrt{p/3}Z$ | Unital | Identity with prob. $1-p$, random Pauli with prob. $p/3$ each | Yes (non-CP) | ||
| Amplitude Damping | $A0 = \begin{bmatrix} 1 & 0 \ 0 & \sqrt{1-\gamma} \end{bmatrix}$, $A1 = \begin{bmatrix} 0 & \sqrt{\gamma} \ 0 & 0 \end{bmatrix}$ | Non-unital | Energy dissipation: $ | 1\rangle \rightarrow | 0\rangle$ with prob. $\gamma$ | Yes (non-CP) |
| Phase Damping | $A0 = \begin{bmatrix} 1 & 0 \ 0 & \sqrt{1-\lambda} \end{bmatrix}$, $A1 = \begin{bmatrix} 0 & 0 \ 0 & \sqrt{\lambda} \end{bmatrix}$ | Unital | Loss of phase coherence without energy loss | Yes (non-CP) |
Comparative Analysis of Noise Channels in Quantum Chemistry
Impact on Quantum Algorithms for Chemistry
Different noise profiles distinctly impact quantum algorithms central to chemistry simulations, such as the Variational Quantum Eigensolver (VQE) and Quantum Phase Estimation (QPE).
Depolarizing noise consistently degrades performance across all circuit depths and error probabilities. Its unital nature introduces uniform randomization that directly corrupts quantum information, making it particularly detrimental to algorithms requiring precise phase relationships [13].
Amplitude damping noise exhibits more complex behavior. Surprisingly, in shallow quantum circuits (depths of 10-15 gates) with small error rates ($p=0.0005$), amplitude damping can actually improve performance in certain quantum machine learning tasks like quantum reservoir computing compared to noiseless reservoirs [13]. This beneficial effect occurs when the fidelity between noisy and noiseless states remains above 0.96 [13].
Phase damping noise primarily destroys phase coherence, disproportionately affecting algorithms reliant on quantum interference patterns. Its impact falls between depolarizing and amplitude damping in severity [13].
Implications for Chemical Accuracy
For quantum chemistry simulations to provide predictive value, they must achieve chemical accuracy—typically defined as an error of 0.0016 hartree (approximately 1 kcal/mol) in energy calculations [17]. Recent experiments on Quantinuum's H2-2 trapped-ion quantum computer utilizing quantum error correction have reached errors of 0.018 hartree for molecular hydrogen—above chemical accuracy but demonstrating meaningful progress [17].
Table 2: Noise Impact on Quantum Chemistry Simulation Metrics
| Performance Metric | Depolarizing Noise | Amplitude Damping Noise | Phase Damping Noise |
|---|---|---|---|
| Ground State Energy Calculation | Consistently detrimental across all error rates | Can be beneficial in shallow circuits with low error rates | Generally detrimental, but less severe than depolarizing |
| Algorithm Trainability | Induces noise-induced barren plateaus | Context-dependent effects; can sometimes improve generalization | Contributes to barren plateaus |
| Quantum Error Correction Overhead | Requires full surface code protection | May leverage bias-tailored codes for more efficient protection | May leverage bias-tailored codes for more efficient protection |
| Achievable Circuit Depth | Severely limited without correction | Moderate limitations; some beneficial effects in NISQ regime | Moderate limitations |
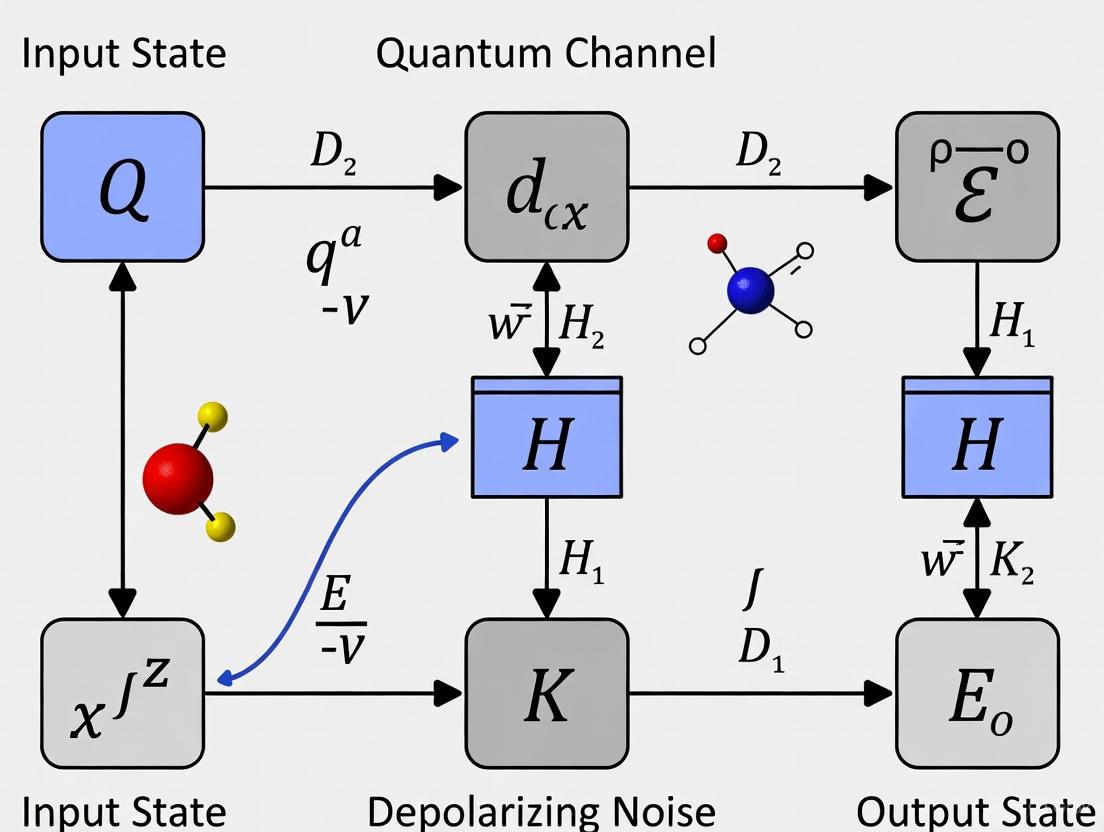
Visualization of the Depolarizing Channel's Effect
The following diagram illustrates the quantum state transformation through a depolarizing noise channel and its relationship to other quantum processes in chemistry simulations:
Experimental Protocols for Noise Characterization and Mitigation
Noise Deconvolution Technique
Noise deconvolution provides a post-processing method to remove known noise effects from measurement statistics. For an observable $O$ measured on a state affected by a known noise channel $\mathcal{E}$, the noise-free expectation value can be estimated as:
$$ \langle O \rangle{\text{mitig}} = c \langle O \rangle{\text{noisy}} $$
where the factor $c$ depends on the specific noise channel and its parameters [15]. This technique applies mathematical inversion of the noise map—which is typically not a completely positive map—as a classical post-processing step rather than a physical reversal.
The depolarizing channel admits an inverse map that can be applied through this deconvolution approach, though it comes at the cost of increased estimation variance: $\operatorname{Var}[\langle O \rangle{\mathrm{mitig}}] \sim c^{2} \operatorname{Var}[\langle O \rangle{\mathrm{noisy}}]$, requiring increased sampling to maintain precision [15].
Quantum Error Correction Approaches
For depolarizing noise in quantum chemistry applications, recent experiments have demonstrated complete quantum chemistry simulations using quantum error correction (QEC). Quantinuum researchers implemented the seven-qubit color code to protect logical qubits, inserting mid-circuit error correction routines during quantum phase estimation calculations for molecular hydrogen [17].
This approach showed improved performance despite increased circuit complexity, challenging the assumption that error correction necessarily adds more noise than it removes in near-term devices. The experiment utilized up to 22 qubits with over 2,000 two-qubit gates, achieving energy estimates within 0.018 hartree of the exact value [17].
Resource-Efficient Alternatives
Given the significant overhead of full quantum error correction, researchers have developed partial fault-tolerant methods that trade off some error protection for lower resource requirements. These include:
- Bias-tailored codes that specifically target the most common error types
- Repetition cat qubits that exploit hardware-level protection against bit-flip errors, reducing the surface code overhead required for fault tolerance [11]
- Dynamic decoupling techniques to suppress memory noise during qubit idling
Table 3: Research Reagent Solutions for Noise Mitigation
| Tool/Technique | Function | Implementation Considerations |
|---|---|---|
| Noise Deconvolution | Classical post-processing to invert known noise channels | Increases measurement variance; requires accurate noise characterization |
| Quantum Error Correction Codes | Active protection of logical qubits using redundancy | High qubit overhead; requires specific connectivity (e.g., surface code, color code) |
| Probabilistic Error Cancellation | Quantum circuit sampling with inverse noise maps | Active mitigation requiring quantum circuit regeneration |
| Cat Qubits | Hardware-level bit-flip suppression using repetition codes | 27× reduction in physical qubits compared to transmons for same logical performance [11] |
| Cluster Expansion Approximation | Systematic noise modeling with controlled accuracy | Honest noise modeling that doesn't underestimate errors [16] |
Depolarizing noise represents a fundamental challenge in quantum computational chemistry due to its uniformly detrimental effect on quantum information. Its mathematical formulation through Kraus operators provides the foundation for developing effective mitigation strategies, including noise deconvolution and quantum error correction. Current research demonstrates that while depolarizing noise consistently degrades algorithm performance, careful characterization and targeted error suppression can extend the computational reach of near-term quantum devices for chemistry applications. The differential impact of various noise channels—particularly the context-dependent effects of amplitude damping versus the uniformly detrimental nature of depolarizing noise—highlights the importance of noise-aware algorithm design and platform-specific optimization in the pursuit of quantum advantage for chemical simulation.
In the realm of quantum computing and simulation, noise presents a fundamental challenge to accurate information processing. Among the various types of quantum noise, amplitude damping (AD) holds particular significance for modeling energy relaxation processes that occur naturally in physical systems, including molecular simulations [18]. This quantum channel provides a rigorous framework for describing the spontaneous emission of energy from an excited state to a ground state, making it indispensable for simulating realistic quantum systems in chemistry and materials science [19].
The particular relevance of amplitude damping stems from its ability to model energy dissipation due to system-environment coupling, a process ubiquitous in molecular systems and quantum devices [18] [20]. Unlike unital noise channels like depolarizing noise, amplitude damping is non-unital, meaning it does not preserve the identity operator, leading to unique effects on quantum states that more accurately reflect physical decay processes [20] [5]. This characteristic makes AD noise especially important for quantum simulations of chemical and biological systems where energy transfer and relaxation are fundamental processes.
Within the context of quantum noise models for chemistry simulations, understanding amplitude damping is crucial for several reasons. First, it enables researchers to model realistic environmental interactions in molecular dynamics simulations. Second, it provides insights into the fundamental limitations of current noisy intermediate-scale quantum (NISQ) devices for computational chemistry [21] [22]. Finally, developing mitigation strategies specifically tailored to amplitude damping noise can enhance the accuracy and reliability of quantum chemistry calculations on emerging quantum hardware [18] [23].
Mathematical Foundation of Amplitude Damping
Kraus Operator Formalism
Amplitude damping is mathematically described as a completely positive trace-preserving (CPTP) map, which can be represented using the Kraus operator formalism [18] [19]. For a single qubit, the AD channel, denoted as â„°_AD, transforms an input state Ï according to:
â„°AD(Ï) = ∑i Ki Ï Ki^â€
where the Kraus operators {Ki} satisfy the completeness relation ∑i Ki^†Ki = I to ensure trace preservation [18] [22]. For the amplitude damping channel, these operators are explicitly defined as:
- K₀ = |0⟩⟨0| + √(1-γ)|1⟩⟨1| = [ \begin{pmatrix} 1 & 0 \ 0 & \sqrt{1-\gamma} \end{pmatrix} ]
- K₠= √γ|0⟩⟨1| = [ \begin{pmatrix} 0 & \sqrt{\gamma} \ 0 & 0 \end{pmatrix} ]
Here, the damping parameter γ ∈ [0,1] represents the probability of energy dissipation from the excited state |1⟩ to the ground state |0⟩ during the noise interval [18]. Physically, γ encapsulates the rate of energy loss to the environment, with γ = 0 corresponding to no damping and γ = 1 representing complete decay to the ground state.
Effect on Quantum States
The action of the amplitude damping channel on a single qubit density matrix Ï = [ \begin{pmatrix} Ï₀₀ & Ï₀₠\ Ïâ‚â‚€ & Ïâ‚â‚ \end{pmatrix} ] transforms it according to:
â„°_AD(Ï) = [ \begin{pmatrix} Ï₀₀ + γÏâ‚â‚ & \sqrt{1-γ}Ï₀₠\ \sqrt{1-γ}Ïâ‚â‚€ & (1-γ)Ïâ‚â‚ \end{pmatrix} ]
This transformation reveals several key characteristics of amplitude damping [19]:
- The population of the excited state (Ïâ‚â‚) decays at a rate γ
- The coherence terms (Ï₀₠and Ïâ‚â‚€) are attenuated by a factor of √(1-γ)
- The ground state population increases as the excited state decays
Unlike phase damping or depolarizing noise, amplitude damping specifically drives the system toward the ground state |0⟩⟨0|, modeling the physical process of energy relaxation that occurs in real quantum systems due to spontaneous emission or interactions with a zero-temperature environment.
Table 1: Comparison of Common Single-Qubit Noise Channels
| Noise Channel | Kraus Operators | Effect on States | Unital/Non-unital | |
|---|---|---|---|---|
| Amplitude Damping | K₀ = [ \begin{pmatrix} 1 & 0 \ 0 & \sqrt{1-\gamma} \end{pmatrix} ], K₠= [ \begin{pmatrix} 0 & \sqrt{\gamma} \ 0 & 0 \end{pmatrix} ] | Energy loss toward | 0⟩ | Non-unital |
| Phase Damping | Kâ‚€ = [ \begin{pmatrix} 1 & 0 \ 0 & \sqrt{1-\lambda} \end{pmatrix} ], Kâ‚ = [ \begin{pmatrix} 0 & 0 \ 0 & \sqrt{\lambda} \end{pmatrix} ] | Loss of phase coherence | Unital | |
| Depolarizing | K₀ = √(1-p)I, K₠= √(p/3)X, K₂ = √(p/3)Y, K₃ = √(p/3)Z | Random Pauli errors | Unital | |
| Bit Flip | K₀ = √(1-p)I, K₠= √(p)X | Bit flip with probability p | Unital |
Physical Interpretation and Relevance to Molecular Systems
Modeling Energy Relaxation
In molecular and quantum chemical systems, amplitude damping provides a crucial model for energy dissipation processes where a quantum system loses energy to its environment. This occurs through various mechanisms, including spontaneous emission, vibrational relaxation, and energy transfer to solvent molecules in solution-phase chemistry [19]. The AD channel effectively captures the fundamental process of a two-level system transitioning from an excited state to a ground state while transferring energy to its surroundings.
The physical significance of amplitude damping in molecular simulations becomes apparent when considering processes such as:
- Electronic transitions in molecules, where excited electrons relax to lower energy states
- Vibrational relaxation, where highly excited vibrational modes dissipate energy to their environment
- Spin relaxation in magnetic resonance applications
- Energy transfer in photosynthetic complexes and other molecular assemblies
In these processes, the damping parameter γ relates to physical quantities such as transition rates and relaxation timescales, which can be derived from Fermi's golden rule or measured experimentally through techniques like time-resolved spectroscopy.
System-Environment Coupling
Amplitude damping naturally arises from modeling a quantum system coupled to a zero-temperature bath, where the environment can accept energy from the system but cannot donate energy back. This represents scenarios such as:
- A molecule coupled to electromagnetic vacuum fluctuations
- A quantum emitter in a cold environment
- Electronic states interacting with phonon baths at low temperature
The Stinespring dilation theorem provides a powerful representation of amplitude damping through an isometric extension, where the system couples to an environmental ancilla [19]. For amplitude damping, this dilation takes the form:
V|0⟩ = |0,0⟩ V|1⟩ = √(1-γ)|0,1⟩ + √(γ)|1,0⟩
This representation shows that when the system starts in the excited state |1⟩, it has a probability γ of transferring its excitation to the environment, resulting in the system ending in the ground state |0⟩ while the environment becomes excited [19].
Figure 1: System-Environment Interaction in Amplitude Damping
Implications for Quantum Chemistry Simulations
Impact on Quantum Algorithms
The presence of amplitude damping noise significantly affects the performance and reliability of quantum algorithms for chemistry simulations. Recent research has revealed several critical implications:
Generation of Magic: Contrary to most noise types that degrade quantum resources, amplitude damping can actually generate or enhance nonstabilizerness (magic) in certain scenarios, unlike depolarizing noise which universally suppresses magic [20]. This has profound implications for fault-tolerant quantum computation, where magic states are essential resources.
Noise-Induced Barren Plateaus: For variational quantum algorithms (VQAs), amplitude damping can contribute to noise-induced barren plateaus (NIBPs), where cost function gradients become exponentially small as circuit depth increases [5]. However, as a Hilbert-Schmidt (HS)-contractive non-unital map, amplitude damping does not necessarily lead to barren plateaus in all scenarios, suggesting it may be less detrimental to VQA trainability than unital noise in some cases [5].
Algorithm-Specific Effects: Different quantum algorithms exhibit varying resilience to amplitude damping noise. For instance, in hybrid quantum neural networks (HQNNs) for image classification, Quanvolutional Neural Networks (QuanNN) demonstrate greater robustness against amplitude damping compared to other architectures like Quantum Convolutional Neural Networks (QCNN) [21].
Resource Requirements and Limitations
The table below summarizes key resource considerations for quantum chemistry simulations under amplitude damping noise:
Table 2: Resource Considerations for Quantum Chemistry Under Amplitude Damping
| Resource Type | Impact of Amplitude Damping | Mitigation Strategies |
|---|---|---|
| Qubit Coherence | Reduces effective coherence time; limits circuit depth | Error mitigation; dynamical decoupling |
| Circuit Fidelity | Exponential decay with circuit depth and γ | Error correction; purification schemes |
| Magic State Resources | Can be generated or enhanced in certain cases | Leverage noise for resource generation |
| Algorithm Trainability | May contribute to noise-induced barren plateaus | Noise-aware optimization; error mitigation |
Mitigation Strategies for Amplitude Damping
Quantum Purification Techniques
Recent advances in quantum error mitigation have led to specialized purification techniques specifically designed for amplitude damping noise. These approaches can substantially enhance the fidelity of affected states or channels while maintaining low resource overhead [18].
The purification method for AD noise employs a circuit-based approach that requires only one or two ancilla qubits in combination with two Clifford gates (Hadamard and controlled-Z gates) [18]. Unlike previous purification methods that required multiple copies of states, this circuit operates on a single noisy state, reducing circuit dimension and thereby decreasing the likelihood of additional noise, while circumventing limitations imposed by the no-cloning theorem [18].
The purification process works by detecting and effectively filtering out noise-induced errors through post-selection based on ancilla measurement outcomes. When successful, this approach produces an actual purified quantum state that can be directly reused in subsequent computational tasks, achieving higher fidelity with respect to the original state [18].
Figure 2: Quantum Purification Circuit for Amplitude Damping
Error Correction and Hardware Solutions
Beyond purification, several other strategies exist for mitigating amplitude damping in quantum chemistry simulations:
Density Matrix Simulations: Using density matrix simulators like Amazon Braket's DM1 allows researchers to model amplitude damping noise explicitly in their simulations, providing more accurate predictions of algorithm performance on real hardware [22]. These simulators support up to 17 qubits and include predefined amplitude damping channels, eliminating the need to manually define Kraus operators [22].
Domain-Specific Software Platforms: Tools like Kvantify Qrunch provide chemistry-specific approaches that improve hardware utilization for molecular simulations, enabling efficient execution across entire processor architectures despite noise limitations [23]. These platforms have demonstrated 3-4× improvement in problem-size capacity compared to standard approaches [23].
Algorithm Selection and Design: Choosing algorithms with inherent robustness to amplitude damping, such as Quanvolutional Neural Networks, can significantly improve results in noisy environments [21]. Additionally, designing custom circuits that account for the specific characteristics of AD noise can enhance performance.
Experimental Protocols and Methodologies
Simulation-Based Noise Characterization
For researchers investigating amplitude damping effects in molecular systems, the following protocol provides a methodology for systematic noise characterization:
Circuit Preparation: Implement the quantum algorithm of interest using a framework that supports noise simulation, such as Amazon Braket, Qiskit, or BlueQubit [24] [22].
Noise Introduction: Incorporate amplitude damping channels after each gate operation or at specified intervals, using the Kraus operator representation with carefully selected γ parameters based on target hardware characteristics or theoretical models.
Density Matrix Simulation: Execute the circuit using a density matrix simulator (e.g., DM1) to track the complete evolution of the quantum state under amplitude damping noise [22].
Measurement and Analysis: Perform measurements on the output state and compare results with noiseless simulations to quantify the impact of amplitude damping on algorithm performance.
Mitigation Application: Implement appropriate error mitigation strategies (purification, error correction, etc.) and reevaluate performance.
Research Reagent Solutions
The table below outlines essential computational tools and their functions for studying amplitude damping in molecular systems:
Table 3: Research Reagent Solutions for Amplitude Damping Studies
| Tool/Platform | Type | Key Function | Applicability to AD Noise |
|---|---|---|---|
| Amazon Braket DM1 | Density matrix simulator | Simulates noisy quantum evolution with predefined channels | Direct support for amplitude damping channel |
| Kvantify Qrunch | Domain-specific quantum platform | Chemistry workflows with improved hardware utilization | Mitigates effects of noise including amplitude damping |
| BlueQubit Simulator | High-performance simulator | Large-scale quantum circuit simulation | Benchmarks algorithm performance under noise |
| NVIDIA cuQuantum | SDK | Accelerated quantum circuit simulation | Enables efficient noise simulation including AD |
Amplitude damping noise represents a fundamental challenge for quantum simulations of molecular systems, directly modeling the energy relaxation processes that occur in real physical systems. Its non-unital character leads to unique effects on quantum algorithms, differing significantly from more commonly studied unital noise channels like depolarizing noise.
The specialized nature of amplitude damping necessitates tailored mitigation approaches, such as the recently developed purification techniques that offer substantial fidelity improvements with minimal resource overhead [18]. Furthermore, the unexpected ability of amplitude damping to generate magic in certain contexts suggests potential opportunities for leveraging, rather than merely mitigating, this noise source [20].
As quantum hardware continues to advance, developing more sophisticated noise models that accurately capture composite noise effects—including combinations of amplitude damping with other decoherence processes—will be essential for realizing the potential of quantum computing in molecular design and drug development. The integration of application-specific error mitigation strategies, such as those implemented in platforms like Kvantify Qrunch, points toward a future where quantum simulations can provide valuable insights for chemical and pharmaceutical research despite the persistent challenge of noise in NISQ-era devices.
This technical guide provides a comparative analysis of how depolarizing and amplitude damping noise models distinctly impact the accuracy of quantum simulations in chemistry. Framed within broader research on quantum noise for chemical simulations, this whitepaper synthesizes recent findings to demonstrate that, contrary to traditional error correction paradigms, amplitude damping noise can be harnessed to enhance performance in specific quantum machine learning (QML) tasks, whereas depolarizing noise is uniformly detrimental. Targeted at researchers, scientists, and drug development professionals, this document presents structured quantitative data, detailed experimental protocols, and essential toolkits to guide the design of noise-resilient quantum computational chemistry experiments.
The simulation of molecular systems is a cornerstone of chemical research with profound implications for drug discovery and materials science. While quantum computers hold the potential to exponentially speed up these simulations by naturally mimicking quantum phenomena, current Noisy Intermediate-Scale Quantum (NISQ) devices are plagued by decoherence and operational errors [13] [7]. These imperfections, or noise, can drastically alter computational outcomes. Notably, not all noise is created equal. The physical nature of a noise model—specifically, whether it is unital (preserving the identity operator, like depolarizing noise) or non-unital (like amplitude damping)—fundamentally dictates its impact on the fidelity of chemical property predictions [13] [25]. Understanding these differences is not merely an academic exercise; it is a critical step toward developing effective error mitigation strategies and leveraging noise as a potential resource for quantum advantage in chemistry.
Theoretical Foundations of Noise Models
In quantum computing, noise processes are formally described by quantum channels, which are mathematical representations of the evolution of an open quantum system. These are often expressed using the operator-sum representation: (\mathcal{E}(\rho) = \sum{k} Ek \rho Ek^{\dagger}), where ({Ek}) are Kraus operators satisfying the completeness condition (\sumk Ek^\dagger E_k = I) [26]. The distinct characteristics of depolarizing and amplitude damping channels are rooted in their unique Kraus operators.
Depolarizing Noise Channel
The depolarizing channel is a unital channel, meaning it preserves the maximally mixed state (I/d). It models a scenario where the quantum state is replaced with a completely random state with probability (p). For a single qubit, it is defined as [13] [26]: [ \mathcal{E}{D}(\rho) = (1 - p) \rho + \frac{p}{3} (X \rho X + Y \rho Y + Z \rho Z) ] Its Kraus operators are (E0 = \sqrt{1-p} I), (E1 = \sqrt{p/3} X), (E2 = \sqrt{p/3} Y), and (E_3 = \sqrt{p/3} Z). This channel symmetrically applies Pauli errors, effectively scrambling quantum information without any preferred direction.
Amplitude Damping Noise Channel
The amplitude damping channel is a non-unital channel that models the dissipation of energy from a quantum system to its environment, analogous to spontaneous emission. For a single qubit, it describes the decay from the excited state (|1\rangle) to the ground state (|0\rangle) [13] [26]. Its Kraus operators are: [ E0 = \begin{bmatrix} 1 & 0 \ 0 & \sqrt{1 - \gamma} \end{bmatrix}, \quad E1 = \begin{bmatrix} 0 & \sqrt{\gamma} \ 0 & 0 \end{bmatrix} ] The corresponding quantum channel is: [ \mathcal{E}{AD}(\rho) = E0 \rho E0^\dagger + E1 \rho E_1^\dagger ] This channel is not symmetric and provides a physical model for energy loss at a finite temperature.
Comparative Mechanisms of Action
The fundamental difference between these channels lies in their symmetry and physical interpretation. Depolarizing noise acts as a symmetric randomizer, uniformly degrading all information in the system. In contrast, amplitude damping noise introduces an asymmetric, structured decay toward the ground state [13]. This structural difference is the origin of their divergent impacts on chemical accuracy, as the latter can sometimes mimic or even enhance certain learning processes in quantum machine learning algorithms [13] [25].
Experimental Protocols for Noise Impact Analysis
To quantitatively assess the impact of these noise models, researchers can employ the following detailed methodology, which is adapted from benchmark studies in quantum reservoir computing (QRC) and quantum machine learning [13] [27].
Quantum Machine Learning Task Definition
- Objective: Predict the first excited electronic energy ((E1)) of the Lithium Hydride (LiH) molecule from its ground state ((|\psi0\rangle)).
- Significance: Quantum chemistry problems, particularly energy calculation, are a fundamental benchmark for QML due to their direct industrial relevance and the exponential scaling of their classical computational cost [13] [28].
- Algorithm: Quantum Reservoir Computing (QRC). A random quantum circuit (the reservoir) processes the input ground state. The resulting quantum state is measured, and these classical measurement outcomes are fed into a simple machine learning model (e.g., linear regression) for the final prediction of (E_1) [13].
- Reservoir Construction: Implement a family of pseudo-random quantum circuits with a variable number of gates (ranging from 10 to 200 in benchmark studies) [13].
- Noise Injection: After the application of each quantum gate in the reservoir, apply a noise channel.
- For depolarizing noise, each gate is followed by (\mathcal{E}{D}(\rho)) with a chosen error probability (p).
- For amplitude damping noise, each gate is followed by (\mathcal{E}{AD}(\rho)) with a damping parameter (\gamma).
- State Evolution: The system's evolution under noise is simulated using a density matrix representation to fully capture mixed-state dynamics [27] [26].
- Metric Calculation: For each noisy circuit, compute the Mean Squared Error (MSE) between the predicted (E_1) and its true value. Concurrently, track the quantum state fidelity between the final noisy state (\rho) and the ideal noiseless state (|\psi\rangle) [13].
Data Analysis and Comparison
- Performance Benchmarking: Plot the MSE against the number of gates for different error probabilities ((p) or (\gamma)) for both noise models.
- Threshold Identification: Determine the regime (number of gates and error probability) where the performance of the noisy reservoir equals or surpasses that of the noiseless reservoir. Studies indicate that for amplitude damping, this occurs when the state fidelity remains above approximately 0.96 [13].
Quantitative Results and Comparative Analysis
Empirical data from rigorous benchmarking reveals a stark contrast in how these noise models affect chemical prediction accuracy.
Table 1: Performance Comparison of Noise Models on LiH Energy Prediction Task [13]
| Noise Model | Impact on Chemical Accuracy (MSE) | Key Characteristic | Performance Relative to Noiseless |
|---|---|---|---|
| Depolarizing | Significant degradation; MSE increases steadily with gate count and error probability. | Unital (symmetric scrambling) | Always worse, even for small error probabilities. |
| Amplitude Damping | Can be beneficial; MSE lower than noiseless for circuits with < ~135 gates at p=0.0005. | Non-unital (energy dissipation) | Can be superior in low-gate, low-error regimes. |
| Phase Damping | Performance degradation, but less severe than depolarizing noise. | Unital (dephasing without energy loss) | Always worse, but fidelity decreases slower than depolarizing. |
Table 2: Fidelity Thresholds for Beneficial Amplitude Damping Noise [13]
| Error Probability (p) | Maximum Beneficial Gate Count | Average Fidelity (vs. Noiseless) |
|---|---|---|
| 0.0001 | Always beneficial or equal in tested range | > 0.99 |
| 0.0005 | ~135 gates | > 0.96 |
| 0.001 | ~60 gates | ~0.96 |
The data shows that the performance of circuits subjected to depolarizing and phase damping noise monotonically decreases as the number of gates increases. In contrast, for amplitude damping noise, there exists a clear crossover point where the noisy reservoir outperforms the noiseless one. This advantage is confined to a regime of shallow to moderate circuit depths (exemplified by the ~135 gate threshold for (p=0.0005)) and high state fidelity (above 0.96) [13]. This regime is highly relevant for NISQ algorithms, which often rely on shallow circuits [13] [7].
The Scientist's Toolkit: Essential Research Reagents and Solutions
To implement the described experimental protocols, researchers require a suite of software and hardware tools capable of emulating quantum systems with high accuracy and configurability.
Table 3: Essential Tools for Quantum Noise Simulation in Chemistry
| Tool Name / Category | Function / Purpose | Key Features for Noise Research |
|---|---|---|
| Qiskit AerSimulator [27] | Quantum circuit emulator with configurable noise models. | Allows injection of depolarizing, amplitude/phase damping, and custom noise models; can be calibrated with backend parameters from real IBM processors. |
| Eviden Qaptiva [27] | High-performance quantum emulator. | Supports deterministic (density matrix) and stochastic (state vector) noise simulation; suitable for detailed analysis of small-to-medium circuits. |
| Paddle Quantum [26] | Quantum machine learning framework. | Built-in functions for common noise channels (bit-flip, phase-flip, amplitude damping, depolarizing); integrates with machine learning pipelines. |
| Chemical Benchmark (e.g., LiH) [13] [28] | A standardized problem for validating quantum simulations. | Provides a ground-truth reference for assessing algorithmic and noise-induced errors in a chemically relevant context. |
| Density Functional Theory (DFT) [28] | Classical computational method for generating training data. | Used to compute accurate ground-state energies for small molecules, which can serve as training data for QML models. |
| Kopsinine | Kopsinine, CAS:559-51-3, MF:C21H26N2O2, MW:338.4 g/mol | Chemical Reagent |
| KPT-6566 | RORγ Inverse Agonist|2-[[4-[[[4-(tert-Butyl)phenyl]sulfonyl]imino]-1-oxo-1,4-dihydro-2-naphthyl]thio]acetic Acid | 2-[[4-[[[4-(tert-Butyl)phenyl]sulfonyl]imino]-1-oxo-1,4-dihydro-2-naphthyl]thio]acetic Acid is a potent RORγ inverse agonist for autoimmune disease research. For Research Use Only. Not for human or veterinary use. |
Implications for Quantum Error Correction and Mitigation
The divergent impacts of depolarizing and amplitude damping noise lead to a critical strategic implication: not all noise requires the same level of mitigation priority. The finding that amplitude damping can be beneficial in specific QML contexts suggests that a nuanced approach to quantum error correction (QEC) is necessary [13] [29].
- Depolarizing and Phase Damping Noise: These should be high-priority targets for correction and mitigation. Since they offer no observable benefit and consistently degrade performance, applying techniques like Zero-Noise Extrapolation (ZNE) and Probabilistic Error Cancellation (PEC) is crucial for maintaining accuracy [13] [30].
- Amplitude Damping Noise: In the regime where it improves performance, aggressively correcting it could be counterproductive. Instead, researchers might focus on leveraging its structure or developing QEC codes that protect against other error types while preserving the potentially useful aspects of amplitude damping [29]. Recent theoretical work on covariant quantum error-correcting codes aims to achieve exactly this: protecting entangled sensors from noise while preserving their sensitivity for metrology and sensing tasks, a concept transferable to quantum simulation [29].
This analysis demonstrates that the impact of quantum noise on chemical accuracy is not monolithic. While depolarizing noise acts as a uniform disruptor that should be prioritized for mitigation, amplitude damping noise presents a more complex profile, with the capacity to enhance performance in specific, shallow-circuit QML applications. For researchers and professionals in drug development and materials science, these findings underscore the importance of noise-aware algorithm design. The future of practical quantum computational chemistry lies not only in suppressing all noise but also in understanding its physical mechanisms to strategically correct its worst effects while potentially co-opting its more structured forms.
The Critical Role of T1 and T2 Times in Chemical Simulation Fidelity
The accurate simulation of chemical systems, such as the calculation of molecular ground-state energies, represents a promising application for near-term quantum computers [31]. However, the practical execution of these algorithms on Noisy Intermediate-Scale Quantum (NISQ) devices is severely constrained by various sources of quantum noise, which disrupt the delicate quantum states necessary for computation [31] [22]. These disruptions cause the quantum information to fade away, a phenomenon known as decoherence, ultimately randomizing or erasing the information within the quantum system [22]. The fight against decoherence is central to quantum computational chemistry, as these errors directly compromise the accuracy of calculated molecular properties like energies and reaction pathways [31].
Among the most critical parameters characterizing decoherence are the T1 and T2 times of a qubit. These timescales dictate how long a quantum state remains viable for computation and are fundamental components of the noise models that determine the fidelity of chemical simulations. This guide explores the role of T1 and T2 within the broader context of quantum noise models, focusing on their distinct effects in depolarizing and amplitude damping channels, and their profound impact on the performance of chemical simulations such as the Variational Quantum Eigensolver (VQE).
Theoretical Foundations of T1 and T2 Relaxation
Defining T1 and T2 Times
Qubits, the fundamental units of quantum information, are inherently fragile and interact uncontrollably with their environment. This interaction leads to decoherence, which is primarily described by two characteristic times:
- T1 Time (Energy Relaxation Time): This is the timescale for a qubit to spontaneously decay from its excited state (|1⟩) to its ground state (|0⟩). It represents the loss of energy to the environment, analogous to energy dissipation in classical systems. The amplitude damping channel is the quantum noise model that mathematically describes this energy loss process [22].
- T2 Time (Phase Relaxation Time): Also known as the dephasing time, T2 quantifies the timescale over which the phase coherence of a quantum superposition is lost. A qubit in a superposition state (α|0⟩ + β|1⟩) will experience a random shifting of the relative phase between |0⟩ and |1⟩, effectively destroying the quantum interference effects that are crucial for quantum computation. Pure dephasing is modeled by the dephasing noise channel.
A key relationship between these parameters is that the total transverse relaxation time T2 is always less than or equal to twice the longitudinal relaxation time T1 (T2 ≤ 2*T1), a constraint arising from the underlying physics of the noise processes.
Mathematical Representation via Quantum Channels
The evolution of a quantum state under noise is not described by unitary gates but by more general operations known as quantum channels. These are completely positive, trace-preserving maps that act on density matrices (Ï), which can represent both pure and mixed quantum states [22].
The Kraus operator formalism provides a mathematical framework to describe these channels. A quantum channel ð’© acts on a density matrix as ð’©(Ï) = ∑ᵢ Káµ¢ Ï Kᵢ†, where the Káµ¢ are Kraus operators satisfying ∑ᵢ Kᵢ†Káµ¢ = I [22].
Amplitude Damping Channel (T1): This channel models energy relaxation. Its Kraus operators are:
- K₀ = [1 0; 0 √(1-γ)] and K₠= [0 √γ; 0 0] where γ = 1 - e^(-t/T1) is the probability of decay after time t. This channel is nonunital, meaning it has a bias that pushes qubits toward a specific state (the ground state |0⟩) [32]. This directional bias distinguishes it from other noise types and, as recent research suggests, can potentially be harnessed as a computational resource [32].
Dephasing Channel (T2-related): This channel models the loss of phase coherence without energy loss. Its Kraus operators are:
- K₀ = √(1-p) I and K₠= √p Z where p is the probability of a phase flip and Z is the Pauli-Z matrix. The relationship between p and T2 is given by p = (1 - e^(-t/T2))/2.
Depolarizing Channel: This is a unital noise model that represents a worst-case scenario where the qubit is replaced by a completely mixed state with probability p. Its Kraus operators are proportional to the Pauli matrices I, X, Y, Z. Unlike amplitude damping, it scrambles the qubit state evenly without any directional bias [32]. It is often used as a simple model to assess the worst-case impact of noise.
The following table summarizes the key characteristics of these primary noise models:
Table 1: Key Quantum Noise Models and Their Characteristics
| Noise Model | Type | Physical Process | Key Kraus Operators | Effect on Qubit State | |
|---|---|---|---|---|---|
| Amplitude Damping | Nonunital [32] | Energy Relaxation (T1) | K₀, K₠(see above) | Population decay to | 0⟩ |
| Dephasing | Unital | Phase Decay (T2) | K₀ ∠I, K₠∠Z | Loss of phase coherence | |
| Depolarizing | Unital [32] | State Scrambling | Kᵢ ∠I, X, Y, Z | Complete randomization |
Impact on Chemical Simulation Algorithms
Vulnerability of Variational Quantum Eigensolver (VQE)
The VQE algorithm is a leading candidate for finding molecular ground-state energies on NISQ devices [31]. It uses a parameterized quantum circuit (ansatz) to prepare a trial state, whose energy is measured and then minimized via a classical optimizer. The algorithm's performance is highly sensitive to noise, which corrupts the prepared quantum state.
Research on simulating VQE for molecules like sodium hydride (NaH) has shown that noise significantly impacts both the estimated energy and the fidelity of the prepared state compared to the true ground state [31]. The choice of ansatz circuit, derived for instance from Unitary Coupled Cluster (UCC) theory, is critical, as deeper and more expressive circuits are more susceptible to decoherence [31].
Comparative Impact of Different Noise Types
The distinct natures of T1- and T2-driven noise lead to different impacts on computational outcomes:
- Amplitude Damping (T1): This noise introduces a systematic bias toward the ground state. In some contexts, this structured, nonunital nature allows for partial adaptation by learning algorithms [30]. Furthermore, its directional bias has been theorized to enable "RESET" protocols that can recycle noisy ancilla qubits, potentially extending computation depth without mid-circuit measurements [32].
- Dephasing (T2): This noise directly attacks quantum superpositions, which are the bedrock of quantum advantage. It rapidly destroys interference effects needed for accurate quantum phase estimation and state preparation in chemical models.
- Depolarizing Noise: As a unital noise, depolarizing noise introduces significant randomness and is often found to cause severe performance degradation in algorithms like Quantum Reinforcement Learning (QRL) [30]. It represents a worst-case scenario that quickly randomizes the quantum state.
Table 2: Comparative Impact of Noise Models on Algorithm Performance
| Algorithm | Impact of Amplitude Damping (T1) | Impact of Dephasing (T2) | Impact of Depolarizing Noise |
|---|---|---|---|
| VQE | Systematic bias in energy estimation; state fidelity loss. | Destruction of phase-critical superpositions; inaccurate energy. | Severe energy inaccuracies; high state infidelity [31]. |
| Quantum Reinforcement Learning (QRL) | Allows for partial adaptation; less severe degradation [30]. | Disrupts learning dynamics and policy convergence. | Significant performance degradation; introduces high randomness [30]. |
| Generic Quantum Circuits | Can be harnessed for reset in specific protocols [32]. | Limits the coherent depth of computation. | Renders circuits classically simulatable beyond a certain depth [33]. |
The following diagram illustrates the logical relationship between noise sources, their physical effects on a qubit, and the subsequent impact on a chemical simulation's output.
Experimental Protocols for Investigating Noise Effects
To robustly benchmark the performance of quantum chemistry algorithms under realistic noise conditions, researchers employ systematic simulation-based methodologies. The workflow below outlines a standard protocol for evaluating the impact of T1, T2, and other noise models on chemical simulation fidelity.
Workflow for Noisy Simulation of Chemical Problems
Detailed Methodology
A comprehensive experiment, as conducted in studies of noisy quantum circuits for computational chemistry, involves several critical stages [31] [30]:
Problem Encoding:
- The molecular Hamiltonian (H(R)) for a target molecule, such as sodium hydride (NaH), is derived under the Born-Oppenheimer approximation [31].
- This fermionic Hamiltonian is transformed into a qubit representation using a mapping like the Jordan-Wigner transformation, resulting in a Hamiltonian expressed as a sum of Pauli strings: H(R) = Σⱼ cⱼ(R) Pⱼ [31].
Ansatz Preparation:
- A parameterized quantum circuit (ansatz) is selected to prepare the trial wavefunction. Common choices for chemical problems are derived from Unitary Coupled Cluster theory, such as UCCSD (Unitary Coupled Cluster with Singles and Doubles) [31].
Noise Model Configuration:
- A quantum simulator capable of emulating NISQ device behavior is configured. This involves defining a custom noise model or using a predefined one.
- Key parameters to set include:
- T1 and T2 times for each qubit, which define the amplitude damping and dephasing channels.
- Gate error probabilities (e.g., for single- and two-qubit gates).
- Measurement error probabilities.
- These parameters can be extracted from hardware calibration data (e.g.,
rigettidevice properties) or set to desired values for a controlled study [22].
Execution and Optimization:
- The VQE algorithm is run iteratively. In each iteration, the quantum circuit is executed on the noisy simulator (with a sufficient number of "shots" or repetitions), and the classical optimizer (e.g., COBYLA or BFGS) adjusts the parameters to minimize the expected energy [31].
Error Mitigation Application:
- Error mitigation techniques are applied to the noisy results to improve accuracy. As explored in noise-resilient quantum learning, these can include [30]:
- Zero-Noise Extrapolation (ZNE): Running the circuit at different noise levels and extrapolating back to the zero-noise limit.
- Probabilistic Error Cancellation (PEC): Using a linear combination of circuit executions to cancel out the effect of known noise channels.
- Adaptive Policy-Guided Error Mitigation (APGEM): Using reward trends from the learning algorithm to adaptively stabilize the training process against noise fluctuations [30].
- Error mitigation techniques are applied to the noisy results to improve accuracy. As explored in noise-resilient quantum learning, these can include [30]:
Table 3: Key Research Tools and Platforms for Noise Simulation Studies
| Tool / Resource | Function | Example in Research |
|---|---|---|
| Density Matrix Simulator | Simulates open quantum systems and general noise channels, representing the state as a density matrix [22]. | Amazon Braket's DM1 simulator is used to simulate predefined noise channels without manually defining Kraus operators [22]. |
| Noise Model Libraries | Predefined implementations of common noise channels (depolarizing, amplitude damping, dephasing). | Qiskit's AerSimulator allows fine-grained control over noise models to emulate NISQ devices [30]. |
| Error Mitigation Frameworks | Software packages implementing ZNE, PEC, and other mitigation techniques. | Hybrid APGEM–ZNE–PEC framework used to boost robustness of Quantum Reinforcement Learning [30]. |
| Classical Optimizers | Algorithms for tuning variational circuit parameters (e.g., COBYLA, BFGS). | Used in VQE to minimize the energy expectation value, with performance affected by noise [31]. |
| Hardware Calibration Data | Experimentally measured qubit properties (T1, T2, gate fidelities). | Informs realistic noise model parameters for simulation; can be sourced from quantum processors like Rigetti's Aspen-M-2 [22]. |
The exploration of noise in quantum chemical simulations is rapidly evolving beyond simple mitigation. A paradigm shift is underway, moving from viewing noise as a purely detrimental force to understanding its nuanced role and potentially leveraging its structure. Key future directions include:
- Harnessing Nonunital Noise: Recent theoretical work from IBM challenges the classical view by demonstrating that nonunital noise, such as amplitude damping, can be actively used to extend computation depth via "RESET" protocols that recycle noisy ancilla qubits [32]. This turns a bug into a potential feature.
- Advanced Error Mitigation: Hybrid frameworks that combine multiple techniques—such as Adaptive Policy-Guided Error Mitigation (APGEM), Zero-Noise Extrapolation (ZNE), and Probabilistic Error Cancellation (PEC)—are showing promise in providing significant resilience across diverse noise models [30].
- Resource-Efficient Simulations: The use of multi-fidelity modeling, which balances computational cost and accuracy by combining simulations of varying quality, is a powerful strategy for optimizing the design of quantum systems, including reactors, and could be adapted for optimizing quantum algorithm parameters under noise [34].
In conclusion, T1 and T2 times are not just hardware metrics; they are fundamental parameters that define the accuracy and feasibility of quantum computational chemistry. Amplitude damping (T1) and dephasing (T2) have distinct and profound impacts on the fidelity of chemical simulations like VQE. While these noise processes currently pose a significant barrier to practical quantum advantage in chemistry, a deeper understanding of their effects—coupled with advanced error mitigation and the potential to harness specific noise properties—is paving the way for more robust and ultimately successful applications of quantum computing to the simulation of matter.
In the pursuit of quantum advantage for computational chemistry, managing quantum noise is a fundamental challenge. Quantum processing units (QPUs) are susceptible to various noise channels that distort calculations, particularly impacting the precise simulation of molecular systems and the prediction of spectroscopic properties [35]. Unlike classical errors, quantum errors include uniquely quantum phenomena like phase flips and amplitude damping, which have no classical analog and can destroy the quantum coherence essential for chemical computations [36]. In the context of chemistry simulations, such as calculating molecular energies or absorption spectra, these errors can lead to inaccurate predictions of reaction pathways, binding energies, and electronic properties. Current-generation noisy intermediate-scale quantum (NISQ) devices operate without full error correction, making the understanding and mitigation of inherent noise channels like phase damping, bit flip, and measurement errors a critical area of research for quantum computational chemistry [22].
Core Noise Channels: Theory and Impact
Mathematical Foundations of Noise Channels
In quantum computing, the evolution of a quantum state is typically described by unitary operations. However, when a quantum system interacts with its environment, this evolution is no longer unitary and must be described using the density matrix formalism and quantum channels [22]. A general quantum channel ( \mathcal{N} ) acting on a density matrix ( \rho ) is mathematically represented by the Kraus decomposition: [ \mathcal{N}(\rho) = \sumi si Ki \rho Ki^\dagger ] where the ( Ki ) are Kraus operators satisfying ( \sumi Ki^\dagger Ki = I ), and ( si ) represents the probability associated with the application of operator ( Ki ) [22]. This formalism provides a comprehensive framework for modeling various types of noise affecting qubits.
Characterization of Key Noise Channels
The following table summarizes the fundamental noise channels relevant to chemical simulations, their Kraus operator representations, and their primary effects on qubit states.
Table 1: Characterization of Key Noise Channels in Quantum Chemical Simulations
| Noise Channel | Kraus Operators | Mathematical Description | Physical Effect on Qubits |
|---|---|---|---|
| Bit Flip [37] [22] | ( K_0 = \sqrt{1-p} \begin{bmatrix} 1 & 0 \ 0 & 1 \end{bmatrix} )K_1 = \sqrt{p} \begin{bmatrix} 0 & 1 \\ 1 & 0 \end{bmatrix} |
( \mathcal{N}(\rho) = (1-p)\rho + p X \rho X ) | Quantum analog of a classical bit error; flips |0⟩ to |1⟩ and vice versa with probability ( p ). |
| Phase Flip [37] | ( K_0 = \sqrt{1-p} \begin{bmatrix} 1 & 0 \ 0 & 1 \end{bmatrix} )K_1 = \sqrt{p} \begin{bmatrix} 1 & 0 \\ 0 & -1 \end{bmatrix} |
( \mathcal{N}(\rho) = (1-p)\rho + p Z \rho Z ) | Alters the relative phase without changing energy probabilities; transforms |+⟩ to |-⟩. |
| Phase Damping [37] | ( K_0 = \begin{bmatrix} 1 & 0 \ 0 & \sqrt{1-\gamma} \end{bmatrix} )K_1 = \begin{bmatrix} 0 & 0 \\ 0 & \sqrt{\gamma} \end{bmatrix} |
Coherent information is lost without energy loss. | Represents a uniquely quantum phenomenon where information is lost without energy loss. |
| Amplitude Damping [37] [20] | ( K_0 = \begin{bmatrix} 1 & 0 \ 0 & \sqrt{1-\gamma} \end{bmatrix} )K_1 = \begin{bmatrix} 0 & \sqrt{\gamma} \\ 0 & 0 \end{bmatrix} |
Models energy dissipation to the environment. | Qubit loses energy to its environment, causing relaxation from |1⟩ to |0⟩. Characterized by relaxation time Tâ‚. |
| Depolarizing [37] | ( K0 = \sqrt{1-p} I ), ( K1 = \sqrt{p/3} X ),( K2 = \sqrt{p/3} Y ), ( K3 = \sqrt{p/3} Z ) | ( \mathcal{N}(\rho) = (1-p)\rho + \frac{p}{3}(X\rho X + Y\rho Y + Z\rho Z) ) | With probability ( p ), the qubit is replaced by a completely mixed state; otherwise, it is left untouched. |
Impact of Noise on Chemical Computations
In chemical simulations, even small error rates can significantly alter computed molecular properties. For instance, in the variational quantum eigensolver (VQE) algorithm used for ground-state energy calculations, phase damping and bit-flip errors can prevent the algorithm from converging to the correct energy landscape [35]. Quantum linear response (qLR) theory, used for calculating spectroscopic properties, is particularly sensitive to noise in the quantum circuit components that construct the Hessian and metric matrices [35]. Research has demonstrated that coherent noise, such as that from imperfect gate calibration, can be particularly detrimental as it can accumulate systematically throughout a circuit, leading to large errors in the final molecular property predictions [35]. Furthermore, unlike depolarizing noise which universally suppresses nonstabilizerness (or "magic"), amplitude damping—a nonunital channel—can paradoxically generate or enhance this quantum resource under certain conditions, which may have implications for the design of noise-resilient quantum algorithms for chemistry [20].
Experimental Protocols for Noise Characterization
Protocol 1: Characterizing Bit Flip and Phase Flip Errors
Objective: To quantitatively measure the bit flip and phase flip error rates on a target qubit used in a molecular simulation.
Materials & Setup:
- Quantum processor or density matrix simulator (e.g., Amazon Braket DM1) [22]
- Single qubit initialized in the |0⟩ state
- Single-qubit gates: Hadamard (H), Pauli-X
Procedure:
- Bit Flip Characterization:
- Initialize the qubit in |0⟩.
- Apply an X-gate to flip the qubit to |1⟩.
- Let the qubit idle for a time ( t ) or apply a sequence of gates to simulate circuit depth.
- Measure the qubit in the computational basis.
- Repeat for ( N ) shots (e.g., 1000) and record the probability of measuring |0⟩, which represents the bit flip error rate ( p_{\text{bit}} ) [37].
- Phase Flip Characterization:
- Initialize the qubit in |0⟩.
- Apply an H-gate to create the |+⟩ = (|0⟩ + |1⟩)/√2 state.
- Let the qubit idle for time ( t ) or apply a sequence of gates.
- Apply another H-gate to convert phase flips into bit flips.
- Measure the qubit in the computational basis.
- Repeat for ( N ) shots and record the probability of measuring |1⟩, which represents the phase flip error rate ( p_{\text{phase}} ) [37].
Data Analysis: The error rates are calculated directly from the measurement statistics. For the bit flip experiment, ( p{\text{bit}} = N0 / N ), where ( N0 ) is the count of |0⟩ measurements after the X-gate. For the phase flip experiment, ( p{\text{phase}} = N1 / N ), where ( N1 ) is the count of |1⟩ measurements.
Protocol 2: Noise Propagation in an Entangled Chemical State
Objective: To evaluate how noise channels corrupt a maximally entangled Bell pair, a state often encountered in quantum chemistry algorithms for active space simulations.
Materials & Setup:
- Two-qubit quantum processor or density matrix simulator
- Two qubits initialized in |00⟩
- Single-qubit Hadamard gate and two-qubit CNOT gate
Procedure:
- Prepare a Bell state:
- Apply H-gate to qubit 0.
- Apply CNOT gate with qubit 0 as control and qubit 1 as target. The ideal state is (|00⟩ + |11⟩)/√2.
- Apply a custom noise model (e.g., asymmetric depolarizing, amplitude damping) to one or both qubits [37] [22].
- Measure both qubits in the computational basis.
- Repeat for ( N ) shots (e.g., 1000) to collect measurement statistics.
Data Analysis: In a noiseless scenario, measurements yield only |00⟩ and |11⟩. The presence of |01⟩ and |10⟩ outcomes indicates bit flip errors. A reduction in the correlation between the two qubits, quantified by the fidelity between the experimental and ideal Bell state density matrix, provides a comprehensive measure of the total noise impact [22].
Figure 1: Workflow for characterizing noise propagation in an entangled state.
For experimentalists and computational scientists working at the intersection of quantum computing and chemistry, the following tools are essential for conducting noise-aware research.
Table 2: Essential Computational Tools for Noise Research in Quantum Chemistry
| Tool Name | Type/Platform | Primary Function in Noise Research |
|---|---|---|
| Amazon Braket DM1 [22] | Cloud-based density matrix simulator | Simulates mixed states and predefined noise channels; crucial for modeling decoherence. |
| Cirq [37] | Python-based SDK | Provides built-in noise channels (BitFlip, PhaseDamp) for building and testing custom noise models. |
| tUCCSD Ansatz [35] | Quantum Algorithm | A parameterized wavefunction ansatz used in VQE; its depth and structure are sensitive to noise. |
| Quantum Linear Response (qLR) [35] | Quantum Algorithm | Framework for excited states and spectra; used to benchmark how noise degrades predictive accuracy. |
| Root Space Decomposition [38] | Mathematical Framework | Aids in classifying noise propagation through quantum systems to inform error correction strategies. |
Mitigation Strategies and Error-Resilient Algorithms
Mitigating the impact of noise is essential for extracting meaningful results from NISQ-era quantum chemistry simulations. A multi-layered approach is typically required.
Table 3: Error Mitigation Techniques for Chemical Simulations on NISQ Devices
| Mitigation Strategy | Description | Use Case in Chemical Simulations |
|---|---|---|
| Error Mitigation | ||
| Zero-Noise Extrapolation (ZNE) [36] | Runs the same circuit at increased noise levels (e.g., by stretching gates) and extrapolates back to the zero-noise result. | Extracting a more accurate ground-state energy from a noisy VQE calculation. |
| Probabilistic Error Cancellation [36] | Uses a known noise model to invert the effect of errors during classical post-processing of results. | Correcting systematic errors in measurement of expectation values for molecular properties. |
| Dynamical Decoupling [36] | Applies rapid sequences of pulses to idle qubits to average out slow noise from the environment. | Protecting encoded quantum information in a molecular wavefunction during periods of idle circuit time. |
| Error Correction | ||
| Surface Codes [36] [39] | A topological QEC code where qubits are arranged in a 2D lattice; only requires local stabilizer measurements. | The leading candidate for protecting logical qubits encoding molecular orbital information in future fault-tolerant QPUs. |
| Shor's Code [36] [39] | The first QECC; uses 9 physical qubits to encode 1 logical qubit and can correct an arbitrary error on any single physical qubit. | A foundational code for demonstrating fault-tolerant quantum memory for chemical state preparation. |
| Ansatz-Based Mitigation [35] | Tailoring the wavefunction ansatz (e.g., in VQE) to reduce circuit depth and susceptibility to noise. | Using an orbital-optimized oo-tUCCSD ansatz to minimize the number of gates needed for an accurate solution. |
Algorithmic Selection and Noise Resilience
The choice of quantum algorithm is critical for noise resilience. For example, the Orbital-Optimized Quantum Linear Response (oo-qLR) method has been identified as a suitable algorithm for the NISQ era because it can be formulated within an active space approximation, reducing the qubit count and circuit depth required [35]. Furthermore, techniques like "Pauli saving" can significantly reduce the number of measurements required for subspace methods like qLR, thereby reducing the cumulative impact of measurement errors [35]. As shown in hardware experiments, even with current error rates, it is possible to obtain absorption spectra for small molecules using a triple-zeta basis set, achieving accuracy comparable to classical multi-configurational methods, provided these mitigation strategies are employed [35].
Figure 2: Logical relationship between noise types and mitigation/correction strategies.
The accurate simulation of molecular systems on quantum computers requires a deep understanding of inherent noise channels such as phase damping, bit flip, and measurement errors. These noise processes introduce significant challenges by degrading the quantum information necessary for calculating molecular energies and spectroscopic properties. While foundational error correction techniques offer a long-term path to fault tolerance, near-term progress in quantum computational chemistry hinges on the co-development of robust error mitigation strategies and noise-aware algorithms, such as oo-qLR. As hardware continues to advance, the systematic characterization and suppression of these noise channels will be indispensable for transitioning quantum chemistry simulations from proof-of-concept demonstrations to impactful tools in fields like drug discovery [35] [40].
Implementing Noise-Aware Quantum Chemistry: From Theory to Practical Drug Discovery Applications
Integrating Noise Models into Variational Quantum Eigensolver (VQE) for Molecular Energy Calculations
The Variational Quantum Eigensolver (VQE) has emerged as a leading hybrid quantum-classical algorithm for calculating molecular ground state energies on Noisy Intermediate-Scale Quantum (NISQ) devices. Its hybrid nature makes it particularly suitable for current hardware limitations. However, the algorithm's performance is critically limited by inherent hardware noise, including quantum decoherence, measurement errors, and gate operation imprecisions [41]. For quantum chemistry simulations to achieve practical utility in fields like drug development and materials science, understanding and integrating these noise models into the computational framework is not merely beneficial—it is essential.
The core objective of VQE in quantum chemistry is to find the ground-state energy (E_0) of a molecular Hamiltonian (H) by minimizing the expectation value (\langle \psi(\boldsymbol{\theta}) | H | \psi(\boldsymbol{\theta}) \rangle) with respect to the parameters (\boldsymbol{\theta}) of a parameterized quantum circuit (ansatz) [41]. On real hardware, this process is disrupted by noise, which can cause significant deviations from the true energy values. The central challenge is that even small error probabilities, on the order of (10^{-4}) to (10^{-2}), can be sufficient to destroy chemical accuracy, typically defined as 1.6 × 10â»Â³ Hartree [42]. This technical guide details the predominant noise models affecting VQE performance and outlines advanced error mitigation strategies necessary for obtaining chemically meaningful results.
Predominant Noise Models in VQE Simulations
Characterization of Common Noise Types
Quantum hardware subjects VQE calculations to several types of noise, each with distinct physical origins and effects on algorithmic performance.
Depolarizing Noise: This model represents a worst-case scenario where a quantum state is completely randomized with probability (p). For a single qubit, the depolarizing channel is described by the map (\epsilon(\rho) = (1-p)\rho + p\frac{I}{2}). Its effect is a uniform degradation of all quantum information [41] [42].
Coherent Noise: Arising from systematic control errors, coherent noise includes miscalibrations in gate operations that lead to over- or under-rotations. Unlike stochastic noise, coherent errors can accumulate constructively, causing substantially larger errors than depolarizing noise with similar strength. These errors are particularly challenging as they do not average out over multiple runs [43].
Amplitude Damping: This noise model captures the energy dissipation effects due to spontaneous emission or interactions with a zero-temperature environment. The Kraus operators for amplitude damping are (E0 = \begin{bmatrix} 1 & 0 \ 0 & \sqrt{1-\gamma} \end{bmatrix}) and (E1 = \begin{bmatrix} 0 & \sqrt{\gamma} \ 0 & 0 \end{bmatrix}), where (\gamma) is the decay probability [41].
Memory Noise: Particularly relevant for trapped-ion and other architectures with long-lived qubits, memory noise refers to errors that accumulate while qubits are idle during computation. Research has identified this as a dominant error source in certain quantum chemistry simulations [17].
Quantitative Impact of Noise on VQE Performance
The following table summarizes key findings from numerical simulations quantifying the impact of depolarizing noise on VQE accuracy across different molecular systems:
Table 1: Maximum Tolerable Depolarizing Gate-Error Probabilities (p_c) for Chemical Accuracy in VQE
| Molecule (Number of Orbitals) | VQE Algorithm Type | p_c (No Error Mitigation) | p_c (With Error Mitigation) |
|---|---|---|---|
| Small molecules (4-14 orbitals) | ADAPT-VQE | (10^{-6}) to (10^{-4}) | (10^{-4}) to (10^{-2}) |
| Small molecules (4-14 orbitals) | Fixed Ansatz (UCCSD, k-UpCCGSD) | Lower than ADAPT-VQE | Lower than ADAPT-VQE |
| Hâ‚‚ | Quantum Phase Estimation with QEC | N/A | Within 0.018 Hartree of exact value [17] |
| Hâ‚„ | Noise-Mitigated VQE [41] | N/A | Errors constrained within (\mathcal{O}(10^{-2}) \sim \mathcal{O}(10^{-1})) |
The scaling relationship between the maximum tolerable gate-error probability (pc) and the number of noisy two-qubit gates (N{\text{II}}) follows (pc \propto N{\text{II}}^{-1}) for any gate-based VQE [42]. This inverse relationship highlights the critical trade-off between algorithmic complexity and noise resilience: as circuit depth increases to model more complex molecules, the hardware must correspondingly improve in fidelity to maintain accuracy.
Advanced Error Mitigation Methodologies
Zero-Noise Extrapolation (ZNE) Enhanced with Neural Networks
Zero-Noise Extrapolation (ZNE) is a powerful error mitigation technique that operates by intentionally increasing the noise level in a controlled manner, measuring the observable of interest at different noise levels, and then extrapolating back to the zero-noise limit.
The standard ZNE protocol involves:
- Noise Scaling: Deliberately increasing the noise level by factors (\lambda > 1) through methods like pulse stretching or identity insertion [41] [43].
- Measurement: Executing the circuit and measuring the energy expectation at each noise factor (\lambda).
- Extrapolation: Fitting the measured data to an exponential or polynomial model and extrapolating to (\lambda = 0).
Recent advancements have integrated neural networks to improve the accuracy of the noise-fitting function. The neural network learns the complex relationship between noisy measurements and the corresponding error-free values, significantly enhancing the extrapolation precision compared to traditional parametric models [41].
Randomized Compiling (RC) for Coherent Noise Conversion
Randomized Compiling (RC) addresses the challenging problem of coherent errors by converting them into stochastic noise, which is more amenable to mitigation through ZNE [43]. The technique works by:
- Circuit Randomization: Decomposing the original circuit into a set of logically equivalent implementations that differ by Pauli twirling operations.
- Random Selection: For each execution, randomly selecting one of these equivalent circuits.
- Averaging: Averaging the results over many randomizations, which effectively transforms coherent errors into stochastic Pauli errors.
The combination of RC and ZNE creates a powerful synergetic effect: RC first converts coherent noise into stochastic noise, which ZNE then effectively extrapolates to the zero-noise limit [43]. Numerical simulations demonstrate that this combination can mitigate energy errors induced by various types of coherent noise by up to two orders of magnitude [43].
Quantum Error Correction (QEC) Integration
While full fault-tolerant quantum computation remains a long-term goal, recent experiments have demonstrated the feasibility of integrating quantum error correction into quantum chemistry simulations. Researchers at Quantinuum implemented the first complete quantum chemistry simulation using quantum error correction on real hardware, calculating the ground-state energy of molecular hydrogen with a seven-qubit color code to protect each logical qubit [17].
Key insights from this demonstration include:
- Mid-circuit correction: Inserting QEC routines during circuit execution, not just at the end, significantly improves results despite increased circuit complexity.
- Partial fault-tolerance: Using bias-tailored codes that focus on correcting the most common error types can provide practical benefits with lower overhead than full fault-tolerance.
- Hardware compatibility: The Hâ‚‚ trapped-ion quantum computer's high-fidelity gates, all-to-all connectivity, and native support for mid-circuit measurements were essential for successful implementation [17].
Algorithm-Specific Noise Resilience Techniques
Beyond general error mitigation methods, algorithm-specific enhancements can significantly improve VQE's noise resilience:
ADAPT-VQE: This iterative ansatz construction method outperforms fixed ansatz approaches (like UCCSD and k-UpCCGSD) in noisy conditions. By growing the circuit iteratively based on energy gradient measurements, ADAPT-VQE achieves comparable accuracy with shallower circuits, reducing noise accumulation [42].
Matrix Product State (MPS)-Inspired Circuits: Designing quantum circuits with reference to MPS structure enables efficient pre-training of parameters on classical computers, ensuring circuit stability and mitigating fluctuations caused by random initialization [41].
Hamiltonian Pauli String Grouping: Intelligent grouping of commuting Pauli terms in the Hamiltonian measurement reduces the number of distinct circuit executions required, thereby decreasing both sampling overhead and measurement error accumulation [41].
Experimental Protocols for Noise-Integrated VQE
Comprehensive Noise-Mitigated VQE Workflow
Diagram: Integrated VQE Workflow with Error Mitigation
The experimental workflow for implementing a comprehensive noise-mitigated VQE involves both quantum and classical components:
Initial State Preparation: Initialize the quantum state as the Hartree-Fock state, which serves as a chemically relevant starting point [41].
Wavefunction Construction and Pre-processing: Construct the wavefunction in matrix product state (MPS) form and gauge it to center-orthogonal form to enhance computational stability and reduce expectation computation complexity [41].
Classical Pre-training: Pre-train the MPS-inspired quantum circuit parameters on classical computers to avoid instability from random initialization and provide a near-optimal starting point for quantum optimization [41].
Parameterized Circuit Execution with Error Mitigation:
Neural Network-Enhanced Zero-Noise Extrapolation: Process the noise-scaled measurements through a neural network trained to model the noise behavior and extrapolate to the zero-noise limit [41].
Classical Optimization: Use the error-mitigated energy estimate to compute gradients and update circuit parameters using classical optimizers like Stochastic Gradient Descent (SGD) [41].
Convergence Check: Repeat steps 4-6 until energy convergence within a predetermined threshold, typically targeting chemical accuracy (1.6×10â»Â³ Hartree).
Protocol for Depolarizing Noise Characterization
To quantitatively characterize the impact of depolarizing noise on VQE performance:
Noise Model Implementation: Implement a depolarizing noise channel after each gate operation in the quantum circuit simulation, with probability parameter (p) [42].
Systematic Scanning: Sweep (p) across a range from (10^{-6}) to (10^{-1}) in logarithmic steps.
Energy Calculation: For each (p) value, execute the full VQE workflow and record the final energy estimate.
Accuracy Threshold Identification: Identify the maximum (p) value at which the energy error remains below the chemical accuracy threshold.
Scaling Analysis: Analyze how this maximum tolerable (p) scales with molecular size and circuit depth, establishing the (pc \propto N{\text{II}}^{-1}) relationship [42].
Coherent Noise Mitigation Protocol
For characterizing and mitigating coherent noise:
Noise Injection: Introduce controlled coherent errors through systematic gate parameter miscalibrations (e.g., consistent over-rotation by angle (\theta)).
Randomized Compiling Application: Implement RC by generating multiple logically equivalent circuit randomizations via Pauli twirling [43].
Sampling and Averaging: Execute each randomized circuit version multiple times and average the results.
ZNE Application: Apply ZNE to the RC-averaged results to further reduce residual stochastic errors [43].
Performance Comparison: Compare the error-mitigated results against both unmitigated results and those obtained using ZNE alone to quantify the synergetic effect.
Table 2: Key Research Reagents and Computational Tools for Noise-Integrated VQE
| Resource/Tool | Type | Function/Role in Noise-Integrated VQE |
|---|---|---|
| MindQuantum | Quantum Computing Framework | Platform for simulating VQE with customizable noise models [41] |
| Matrix Product States (MPS) | Classical Simulation Tool | Provides pre-training capability for stable circuit initialization [41] |
| Seven-Qubit Color Code | Quantum Error Correction Code | Protects logical qubits in fault-tolerant chemistry simulations [17] |
| Randomized Compiling (RC) | Software Technique | Converts coherent noise into stochastic noise for improved mitigation [43] |
| Neural Network ZNE Fitting | Machine Learning Component | Enhances accuracy of zero-noise extrapolation through learned noise models [41] |
| Depolarizing Noise Model | Noise Characterization Tool | Benchmarks worst-case performance under uniform stochastic noise [42] |
| Grouped Pauli Measurements | Measurement Protocol | Reduces sampling overhead and measurement error accumulation [41] |
| ADAPT-VQE Algorithm | Quantum Algorithm | Iterative ansatz construction for reduced circuit depth and noise resilience [42] |
The integration of noise models into VQE for molecular energy calculations has evolved from a theoretical consideration to an essential component of practical quantum computational chemistry. The methodologies outlined in this guide—including Zero-Noise Extrapolation enhanced with neural networks, Randomized Compiling, and algorithm-specific optimizations—represent the current state-of-the-art in mitigating hardware noise.
The quantitative relationship (pc \propto N{\text{II}}^{-1}) establishes a clear benchmark for hardware development: as researchers target larger molecular systems requiring deeper circuits, simultaneous improvements in gate fidelities are imperative [42]. The demonstration that error correction can improve quantum chemistry calculations despite increased circuit complexity marks a significant milestone toward fault-tolerant quantum chemistry simulations [17].
For researchers in drug development and materials science, these noise integration and mitigation strategies make quantum computational chemistry increasingly relevant for practical applications. As hardware continues to improve and error mitigation strategies become more sophisticated, the prospect of achieving quantum advantage for real-world chemical problems appears increasingly attainable. Future research directions will likely focus on optimizing the resource trade-offs between error correction, mitigation, and algorithmic efficiency to maximize the computational power of near-term quantum devices for chemical discovery.
Noise-Adaptive Compilation Strategies for Chemical Circuit Optimization
In the Noisy Intermediate-Scale Quantum (NISQ) era, simulating chemical systems such as hydrogen chains presents a significant challenge due to the susceptibility of quantum processors to inherent noise [44]. Variational Quantum Algorithms (VQAs), particularly the Variational Quantum Eigensolver (VQE), have emerged as a primary framework for tackling quantum chemistry problems. However, their performance is severely limited by quantum decoherence, gate infidelities, and the notorious noise-induced barren plateaus (NIBPs) phenomenon, where gradients of cost functions become exponentially small, crippling algorithm trainability [5].
Compilation—the process of transforming high-level quantum algorithms into hardware-executable instructions—is a critical determinant of computational fidelity. Standard compilers often map logical circuits directly to the entire physical device, ignoring the non-uniform spatial distribution of hardware noise. This oversight can lead to circuits being deployed on high-error qubits and links, drastically reducing result accuracy [45]. Noise-adaptive compilation strategies proactively address these limitations by leveraging real-time hardware calibration data and noise models to guide the translation process, thereby significantly enhancing the robustness and accuracy of chemical simulations on near-term quantum hardware. This technical guide provides a comprehensive overview of these strategies, framed within the context of chemical circuit optimization.
Quantum Noise Models in Chemistry Simulations
Accurately modeling quantum noise is the cornerstone of developing effective noise-adaptive compilers. Different noise channels affect quantum circuits in distinct ways, and their impact is particularly pronounced in the deep circuits required for chemistry simulations like the Unitary Coupled Cluster (UCC) ansatz.
Table 1: Common Quantum Noise Models and Their Impact on Chemical Simulations
| Noise Model | Physical Cause | Key Mathematical Formulation | Impact on VQE for Chemistry |
|---|---|---|---|
| Depolarizing Channel | Unintended interactions with the environment | Replaces qubit state with max. mixed state (prob. ( p )) | Causes severe performance degradation; major contributor to NIBPs [30] [5]. |
| Amplitude Damping | Energy dissipation (Tâ‚ relaxation) | Kraus operators: ( E0 = \begin{bmatrix} 1 & 0 \ 0 & \sqrt{1-\gamma} \end{bmatrix}, E1 = \begin{bmatrix} 0 & \sqrt{\gamma} \ 0 & 0 \end{bmatrix} ) | Represents energy loss; is HS-contractive non-unital; can lead to NILS [5]. |
| Phase Damping | Loss of phase coherence (Tâ‚‚ relaxation) | Kraus operators diagonal | Causes pure dephasing; investigated alongside amplitude damping in scaling studies [44]. |
| Bit/Phase Flip | Control errors or environmental interactions | Pauli X (Bit Flip) or Pauli Z (Phase Flip) applied with probability ( p ) | Disrupts quantum state; part of comprehensive noise robustness evaluations [21]. |
For chemical simulations, the interplay between circuit depth and these noise models is critical. Scaling studies of VQE for hydrogen chains using the UCC ansatz have focused on how noise channels like depolarization, amplitude damping, and phase damping affect gradients and optimizability as the system size grows towards 28 qubits [44]. Furthermore, non-unital noise models like amplitude damping can lead not only to NIBPs but also to noise-induced limit sets (NILS), where the cost function converges to a range of values instead of a single minimum, further complicating the optimization landscape [5].
Core Noise-Adaptive Compilation Strategies
Resource Virtualization and Subregion Selection
The QSteed compilation system introduces a paradigm of resource virtualization and a select-then-compile workflow, which is highly effective for chemical circuits [45]. Instead of mapping a circuit to an entire quantum chip, the compiler virtualizes the physical hardware into a hierarchy of abstractions: the real Quantum Processing Unit (QPU), a standard QPU (StdQPU), a substructure of the QPU (SubQPU), and Virtual QPUs (VQPUs). These VQPUs represent high-quality, low-noise subregions of the larger chip identified using heuristic strategies and real calibration data.
The operational workflow, crucial for executing a chemical VQE circuit, is as follows:
- The input quantum circuit (e.g., a UCC ansatz) is standardized into a hardware-agnostic representation.
- The compiler queries a pre-computed resource database to select the optimal VQPU that best matches the circuit's structural topology or offers the highest predicted fidelity.
- All subsequent compilation steps—including layout, routing, and gate resynthesis—are confined to this selected VQPU.
- The final, hardware-executable quantum assembly language (QASM) code is generated and verified.
This strategy demonstrably outperforms direct full-chip mapping, achieving higher circuit fidelity by concentrating computation on the most reliable hardware resources [45].
Figure 1: The select-then-compile workflow for noise-adaptive quantum compilation, as implemented in the QSteed architecture [45].
Noise-Adaptive Transpilation and Mapping
A pivotal finding in noise-adaptive compilation is that simply concentrating a workload onto a small subset of the "best" qubits can sometimes increase error variability due to increased load and crosstalk [46]. This insight has led to the development of sophisticated noise-adaptive transpilation and mapping techniques. One such approach is Noise-Directed Adaptive Remapping (NDAR), which dynamically remaps logical qubits to physical qubits during the optimization process based on the characteristics of noisy output samples [47].
The NDAR process, which can be integrated into hybrid quantum-classical algorithms like VQE, involves:
- Sample Generation: Obtain a set of samples (e.g., molecular energy measurements) from a noisy quantum device.
- Problem Adaptation: Analyze the samples to identify an "attractor state" or consensus bitstring. This information is used to apply a gauge transformation (e.g., a bit-flip) to the problem Hamiltonian, effectively guiding the search away from noisy attractors.
- Re-optimization: Resolve the transformed optimization problem on the quantum processor. This iterative process allows the algorithm to exploit, rather than be defeated by, the noise present in the system. Enhancements to NDAR include incorporating delay-gate-induced amplitude damping to better model and adapt to realistic noise patterns [47].
Integrated Error Mitigation at the Compiler Level
For chemistry simulations, combining noise-adaptive compilation with circuit-level error mitigation techniques creates a powerful synergy. While compilation strategies proactively avoid noise, error mitigation techniques suppress the impact of the noise that remains.
Table 2: Error Mitigation Techniques Integrable with Noise-Adaptive Compilers
| Technique | Methodology | Experimental Protocol for Chemistry VQE | Compiler Consideration |
|---|---|---|---|
| Zero-Noise Extrapolation (ZNE) | Intentionally scales noise by stretching pulses/inserting gates, then extrapolates to zero-noise. | Execute the same UCC circuit at 1.0x, 1.5x, and 2.0x base depth; use linear/exp. extrapolation on measured energies [30]. | Compiler must support gate insertion for noise scaling without disrupting topology. |
| Probabilistic Error Cancellation (PEC) | Constructs noisy circuit as a linear combination of executable circuits to approximate noiseless output. | For each native gate, pre-characterize its response to noise. In runtime, sample from a tailored "quasi-probability" distribution of circuits [30]. | Significant overhead; compiler must manage complex circuit decomposition and high sampling count. |
| Measurement Error Mitigation | Pre-characterizes readout errors via tensorably of calibration matrices and applies inverse during post-processing. | Prepare and measure all computational basis states for the active qubit register; construct a response matrix ( M ); correct results by applying ( M^{-1} ) [30]. | A standalone post-processing step; requires compiler to allocate time for calibration cycle. |
A robust approach for chemical simulations is to deploy a hybrid error mitigation framework. For instance, one can synergistically integrate Adaptive Policy-Guided Error Mitigation (APGEM) with ZNE and PEC. This combination has been shown to significantly improve convergence stability, solution quality (approximation ratio), and informational coherence (fidelity) for quantum algorithms under realistic noise conditions, as demonstrated in solving problems like the Traveling Salesman Problem [30].
Experimental Protocols for Benchmarking
To validate and compare the efficacy of different noise-adaptive compilation strategies for chemical simulations, a rigorous and standardized experimental protocol is essential.
Protocol 1: VQE for Hydrogen Chain Energy Estimation
This protocol is designed to evaluate how compilation strategies affect the accuracy and trainability of a concrete chemical problem.
- Objective: Compute the ground state energy of a linear hydrogen chain (Hâ‚„ to Hâ‚â‚‚) using the VQE algorithm with a UCC ansatz, and compare the result to the Full Configuration Interaction (FCI) benchmark.
- Testbed: Simulated quantum environment (e.g., Qiskit AerSimulator) configured with real backend calibration data (e.g., from IBM's
ibm_perthor Quafu'sBaihuaprocessor) and programmable noise models [27]. - Methodology:
- Circuit Synthesis: Generate the parameterized UCC ansatz circuit for the target molecule.
- Compilation Strategies:
- Baseline: Compile with a standard compiler (e.g., Qiskit without noise-adaptive settings).
- Test: Compile with the noise-adaptive strategy under test (e.g., QSteed's VQPU selection or an NDAR-integrated workflow).
- Execution: Run the VQE optimization loop (e.g., using the L-BFGS-B optimizer) for both compiled circuits on the noisy simulator.
- Metrics:
- Energy Error: Final VQE energy vs. FCI energy (Ha).
- Approximation Ratio: (EVQE - Emax) / (EFCI - Emax).
- Circuit Fidelity: State fidelity between the noisy output state and the ideal state.
- Gradient Norms: Monitor for signs of NIBPs during optimization.
Protocol 2: Cross-Platform Fidelity Deviation Assessment
This protocol assesses how well a noise-adaptive compilation pipeline can replicate real hardware behavior across different simulation environments, which is crucial for pre-fabrication testing and algorithm development.
- Objective: Quantify the fidelity deviation between a real quantum backend and various quantum emulators (e.g., Qaptiva, Qiskit Aer) when using noise-adaptive compilation.
- Testbed: Real quantum device (e.g., IBM's 7-qubit
ibm_perth) and its emulated counterparts [27]. - Methodology:
- Noise Model Calibration: For the emulators, configure noise models (Depolarizing, Thermal Relaxation, Amplitude Damping) using the latest calibration parameters (T1, T2, gate error rates) from the real backend.
- Circuit Benchmarking: Execute a suite of benchmark circuits, including:
- Randomized Benchmarking Circuits: For gate-level fidelity.
- Small Chemistry Circuits: Such as VQE for Hâ‚‚ or HeHâº.
- Metric Calculation: For each circuit, compute the fidelity deviation: ( \Delta F = |F{\text{emulator}} - F{\text{real device}}| ).
- Analysis: Document the runtime and software/hardware flexibility of each emulation environment. A successful noise-adaptive setup should achieve a low ( \Delta F ) (e.g., <1% for Qaptiva in deterministic mode [27]).
The Scientist's Toolkit
Table 3: Essential Research Reagents and Tools for Noise-Adaptive Compilation Research
| Tool/Solution | Type | Primary Function in Research | Example/Reference |
|---|---|---|---|
| Qiskit AerSimulator | Quantum Emulator | Provides a configurable simulation backend for testing noise models and compilation strategies without requiring real QPU access [27]. | AerSimulator(method='automatic', noise_model=noise_model) |
| Eviden Qaptiva | Quantum Emulator | Offers high-accuracy, deterministic emulation of small quantum systems with noise, useful for precise validation of noise models [27]. | Qaptiva 802 with deterministic mode (~20 physical qubits) |
| Calibration Data | Dataset | Provides real-time parameters (gate errors, T1, T2) from a quantum processor; essential for constructing accurate noise models. | IBM's backend.properties() or Quafu's calibration data feed [45] [27] |
| ZX-Calculus / Tensor Networks | Circuit Representation | Alternative representations that can simplify circuit structures and expose optimization opportunities not visible in the gate model [48]. | Used in synthesis and optimization steps of the compiler [48] |
| Hardware-Efficient Ansatz | Algorithmic Component | A parameterized circuit constructed from a hardware's native gate set, minimizing compilation overhead and depth, thus reducing noise accumulation. | Commonly used as a alternative to UCC in VQE experiments [48] |
| L-669083 | L-669083, CAS:130007-52-2, MF:C29H29IN4O5S, MW:672.5 g/mol | Chemical Reagent | Bench Chemicals |
| KU-0060648 | KU-0060648, CAS:881375-00-4, MF:C33H34N4O4S, MW:582.7 g/mol | Chemical Reagent | Bench Chemicals |
Figure 2: A typical experimental workflow for evaluating noise-adaptive compilation for a chemical VQE simulation.
The pursuit of practical quantum chemistry simulations on NISQ-era hardware necessitates a holistic approach where algorithm design and compilation are deeply intertwined with noise awareness. Noise-adaptive compilation strategies—ranging from resource virtualization and intelligent qubit mapping to the integration of error mitigation techniques—provide a critical pathway to enhancing the fidelity and reliability of these computations. Empirical studies challenge the simplistic view of always using the "best" qubits and highlight the need for sophisticated, dynamic approaches like NDAR. As quantum hardware continues to evolve, the co-design of chemical algorithms and noise-adaptive compilers will remain a vital research frontier, pushing the boundaries of what is possible in computational chemistry and drug development on quantum processors.
Machine Learning Approaches for Data-Efficient Quantum Noise Modeling in Chemical Systems
The accurate simulation of molecules and chemical reactions is a paramount application for quantum computing, as these processes are governed by quantum mechanics. However, the practical utility of current Noisy Intermediate-Scale Quantum (NISQ) devices is severely constrained by various noise mechanisms that hinder algorithmic performance. For chemical systems, where precise energy calculations are critical for predicting reaction pathways and molecular properties, these errors can render simulation results meaningless. Quantum noise introduces decoherence, gate infidelities, and measurement errors that accumulate throughout circuit executions, particularly problematic for chemistry algorithms like the Variational Quantum Eigensolver (VQE) which require numerous iterative measurements to determine molecular ground states.
The core challenge lies in the trade-off between noise model accuracy and experimental characterization overhead. Traditional noise models, often derived from routine device calibration data, typically assume independent Pauli noise channels that fail to capture the complex, correlated error dynamics of real hardware. These models exclude crucial effects such as cross-talk, non-Markovian memory effects, and time-varying decoherence parameters. While comprehensive characterization techniques like quantum process tomography can capture these intricate effects, they are resource-intensive and impractical for routine calibration of larger quantum processors. This creates a critical bottleneck for quantum chemistry applications, where reliable results depend on accurate noise characterization.
Core Machine Learning Framework for Noise Modeling
Parameterized Noise Model Architecture
The data-efficient machine learning framework introduces a circuit-size-independent parameterized noise model that achieves high fidelity by leveraging readily available experimental data. This approach refines an initial parameterized noise model (\mathcal{N}(\bm{\theta})) through an iterative, machine learning-driven optimization process, where the objective is to find optimal parameters (\bm{\theta^*}) that minimize the discrepancy between simulated and experimental output distributions [49] [50].
For single-qubit gates, the error channel (\mathcal{E}g) is modeled as a composition of distinct error channels targeting specific physical mechanisms. The model incorporates thermal relaxation to account for energy relaxation ((T1)) and dephasing ((T_2)) processes occurring during gate operation, combined with a depolarizing component whose parameters are learned rather than fixed by conventional characterization data [50]. This composite structure enables the model to capture more complex error dynamics than standard approaches.
For multi-qubit gates, the framework extends to include correlated error components such as cross-talk, which is particularly important for chemical simulations where entangling gates between qubits are essential for representing electron correlations. The parameterization allows the model to capture spatial and temporal correlations that are typically omitted from standard vendor-provided noise models [50]. The complete model is trained using Gaussian process regression to construct surrogate models of the noise, effectively interpolating between sparsely sampled data points and extrapolating to unmeasured regions of the parameter space [51].
Data-Efficient Training Methodology
The training process leverages a fundamentally different approach from conventional characterization protocols. Rather than requiring dedicated quantum tomography experiments, the framework repurposes data from any existing circuit executions, including application benchmarks, algorithm runs (such as VQE for small molecules), and routine calibration measurements [49] [50]. This eliminates the overhead of specialized characterization protocols and allows the noise model to be tailored specifically to the context of executed quantum chemistry algorithms.
The optimization objective is to minimize the Hellinger distance between the simulated output distribution from the parameterized noise model and the experimental measurement outcomes [50]. This metric quantifies the statistical divergence between probability distributions and provides a robust measure of model fidelity. Through Bayesian optimization of hyperparameters, the framework efficiently explores the parameter space to identify optimal noise model configurations that maximize agreement with experimental data [51].
A crucial feature of this approach is its demonstrated ability to learn from small-scale circuits and generalize to larger systems. Research has shown that models trained exclusively on 4–6 qubit circuits can accurately predict the behavior of larger validation circuits comprising 7–9 qubits [49]. This capability is particularly valuable for chemical systems, where scaling simulations to larger molecules is essential for practical applications.
Table 1: Key Performance Metrics of ML-Based Noise Modeling Framework
| Metric | Standard Noise Model | ML-Based Framework | Improvement |
|---|---|---|---|
| Model Fidelity (Hellinger Distance) | Baseline | Up to 65% reduction | 65% improvement [49] [51] |
| Training Data Requirements | Dedicated characterization circuits | Existing circuit data | Eliminates characterization overhead [50] |
| Generalization Capability | Limited to characterized systems | Accurate prediction for larger circuits | Enables scaling [49] |
| Cross-Platform Adaptability | Device-specific | Validated across multiple IBM processors | Broad applicability [50] |
Experimental Protocols and Validation Methodologies
Benchmarking for Quantum Chemical Applications
The validation of machine learning-derived noise models for chemical systems requires specialized benchmarking protocols. Researchers have employed a multi-faceted approach combining theoretical modeling, numerical simulations, and experimental validation on actual quantum hardware [51]. For quantum chemistry specifically, the framework is tested on algorithms like VQE and the Quantum Approximate Optimization Algorithm (QAOA) applied to molecular systems [50].
The experimental protocol typically involves several key stages. First, an initial training dataset is collected using existing benchmark circuits, which may include simple chemistry-inspired ansatze or textbook algorithms adapted for molecular simulation. For chemical applications, these often involve circuits that prepare model molecular states or simulate basic quantum chemical processes. The measurement outcomes from these circuits are used as the ground truth for optimizing the noise model parameters [49] [50].
Next, the trained model is validated on out-of-distribution circuits, including both generic quantum circuits and specifically chemistry-oriented circuits such as those for estimating molecular energies. Comprehensive benchmarking across multiple quantum devices has demonstrated that this approach delivers a substantial improvement in model fidelity, reducing the mean Hellinger distance to experimental results by 50% with peak reduction of 65% compared to conventional noise models [49] [51].
Integration with Error Mitigation Techniques
For practical quantum chemistry applications, the machine learning-derived noise models can be integrated with advanced error mitigation strategies to further enhance computational accuracy. Research has shown that combining accurate noise modeling with techniques like Zero Noise Extrapolation (ZNE), Probabilistic Error Cancellation (PEC), and measurement error mitigation leads to substantial improvements in algorithm performance [30] [51].
In one systematic study, a hybrid framework integrating Adaptive Policy-Guided Error Mitigation (APGEM) with ZNE and PEC demonstrated significant resilience against various noise types including depolarizing and amplitude damping noise [30]. This is particularly relevant for chemical simulations, where maintaining coherence throughout complex quantum circuits is essential for accurate energy calculations. The adaptive component monitors reward trends (such as energy convergence in VQE) and stabilizes learning under noise fluctuations, making it particularly suitable for the iterative optimization processes central to variational quantum algorithms in chemistry.
Table 2: Experimental Validation Framework for Quantum Noise Models in Chemical Systems
| Validation Component | Implementation in Chemical Context | Key Outcome Measures |
|---|---|---|
| Algorithm Benchmarking | VQE for small molecules (e.g., LiH, Hâ‚‚) | Ground state energy accuracy [28] [7] |
| Noise Resilience Testing | Subjecting circuits to depolarizing, amplitude damping noise | Circuit fidelity, convergence stability [30] |
| Scalability Assessment | Progressive increase in molecular complexity | Generalization to larger active spaces [49] |
| Mitigation Integration | ZNE, PEC applied to chemistry circuits | Error reduction in energy measurements [30] |
Implementation Workflow for Chemical Systems
The following diagram illustrates the complete workflow for implementing machine learning-based quantum noise modeling specifically for chemical simulation applications:
ML Noise Modeling Workflow
Table 3: Research Reagent Solutions for Quantum Noise Modeling Experiments
| Resource Category | Specific Tools & Techniques | Function in Noise Modeling |
|---|---|---|
| Quantum Hardware Platforms | IBM Quantum processors (superconducting), Trapped-ion quantum computers | Provide experimental testbeds for data collection and model validation [50] [52] |
| Software Frameworks | Qiskit, TKET, Munich Quantum Toolkit | Enable circuit construction, simulation, and noise-aware compilation [50] |
| Machine Learning Libraries | Gaussian process regression, Bayesian optimization | Implement surrogate modeling and efficient parameter space exploration [51] |
| Characterization Protocols | Randomized benchmarking, quantum process tomography | Generate ground truth data for model training and validation [50] |
| Error Mitigation Techniques | ZNE, PEC, measurement error mitigation | Enhance result quality in conjunction with accurate noise models [30] |
| Chemical Simulation Tools | VQE, QAOA implementations for molecules | Provide application-specific test cases for noise model validation [50] [7] |
Impact on Quantum Chemistry Applications
Enabling Complex Molecular Simulations
Accurate noise modeling directly enhances the feasibility of simulating chemically relevant molecules. Recent research has demonstrated quantum simulations of molecular dynamics, such as modeling how specific real molecules behave after absorbing light [52]. Using a trapped-ion quantum computer with a mixed qudit-boson simulation technique, researchers simulated the behavior of allene, butatriene, and pyrazine molecules undergoing photochemical processes. This approach was estimated to be at least a million times more resource-efficient than standard quantum approaches, highlighting how tailored noise modeling and simulation techniques can extend the reach of current quantum hardware for chemical applications [52].
For industrial applications, molecules like cytochrome P450 enzymes and the iron-molybdenum cofactor (FeMoco) represent benchmark targets. Understanding P450 interactions with drug candidates at the molecular level helps predict metabolic stability and potential drug-drug interactions, while FeMoco studies could revolutionize fertilizer production through improved understanding of biological nitrogen fixation [11]. These complex systems require sophisticated simulation approaches that account for noise characteristics to produce reliable results.
Resource Estimation and Scalability
The machine learning approach to noise modeling has important implications for resource estimation in quantum chemistry applications. By providing more accurate predictions of how algorithms will perform on real hardware, researchers can make better judgments about the qubit requirements for specific chemical simulations. Recent analyses suggest that with fault-tolerant quantum computers based on advanced qubit technologies like cat qubits, simulating complex molecules such as FeMoco and P450 might require approximately 99,000 physical qubits - a 27-fold reduction compared to estimates for conventional superconducting architectures [11].
The following diagram illustrates the interaction between noise modeling, resource requirements, and chemical application complexity:
Noise Model Impact Pathway
Future Directions and Research Challenges
The development of machine learning approaches for quantum noise modeling in chemical systems continues to face several important challenges. Future research directions include exploring more complex noise models that capture non-Markovian dynamics and time-varying noise characteristics, developing adaptive error mitigation techniques that respond to changing noise conditions, and investigating the scalability of these approaches to larger quantum systems relevant for industrial chemical applications [51].
Integration of noise modeling with hardware-aware algorithm design represents another promising direction. As noted in recent research, "practitioners can leverage their existing experimental datasets to continuously refine noise models, enhancing the development of noise-aware compilation and error mitigation protocols without incurring additional experimental costs" [50]. This continuous refinement cycle will be particularly important for chemical simulations, where different molecular systems and algorithmic approaches may exhibit varying sensitivities to specific noise types.
For the quantum chemistry community, these advances in data-efficient noise modeling create a pathway toward more reliable simulation of complex molecular systems. With accurate noise models, researchers can make better predictions about when quantum computers might achieve practical utility for industrial chemical problems, from drug design and catalyst development to materials science and renewable energy research. As hardware continues to improve, the integration of machine learning-based noise characterization with application-specific optimization will play a crucial role in realizing the potential of quantum computing for chemistry.
The accurate computational modeling of molecular ground states is a cornerstone of modern chemistry, with profound implications for drug discovery and materials science. While classical computational methods, notably Density Functional Theory (DFT), provide high accuracy, they are notoriously resource-intensive, making the simulation of large, scientifically relevant molecular systems prohibitive [53]. The emergence of quantum computing offers a promising alternative, potentially capable of solving these classically intractable problems. Specifically, variational quantum algorithms (VQAs) like the Variational Quantum Eigensolver (VQE) are designed to leverage near-term quantum devices to estimate molecular energies [54].
However, the practical execution of these algorithms on current Noisy Intermediate-Scale Quantum (NISQ) hardware is severely hampered by the pervasive presence of quantum noise. Gate infidelities, decoherence, and measurement errors corrupt quantum information, degrading computational accuracy and algorithm performance [30] [5]. This case study analyzes the impact of realistic noise conditions on molecular ground state modeling, using the BODIPY molecule and the Transverse-Field Ising Model as benchmarks. We provide a quantitative analysis of various noise models, detail robust experimental protocols for high-precision measurement and state preparation, and present a toolkit of error mitigation strategies essential for extracting reliable chemical predictions from today's quantum hardware.
Quantum Noise Models and Their Chemical Impact
Realistic noise simulation requires modeling distinct physical processes, each with a characteristic effect on quantum computation. The performance of quantum algorithms is not uniformly degraded by all noise types; understanding their individual and combined impacts is crucial for developing effective error mitigation strategies.
Table 1: Characteristics and Chemical Impact of Common Quantum Noise Models
| Noise Model | Physical Cause | Primary Effect on Quantum State | Impact on Molecular Energy Estimation | |
|---|---|---|---|---|
| Depolarizing Noise | Uncontrolled interaction with environment | Randomizes the quantum state (complete mixing) | Severe performance degradation; introduces significant randomness [30] | |
| Amplitude Damping | Energy dissipation (e.g., spontaneous emission) | Loss of energy, drives system to | g> ground state | Allows for partial algorithm adaptation; less detrimental than unital noise [30] [5] |
| Dephasing Noise | Elastic scattering causes loss of phase coherence | Suppresses off-diagonal elements in density matrix | Disrupts interference effects crucial for quantum algorithms | |
| Measurement Noise | Inaccurate qubit readout | Bit-flip errors on measurement outcomes | Comparatively milder effect, primarily impacts readout stage [30] |
The BODIPY molecule (Boron-dipyrromethene), a fluorescent dye used in biolabeling and photovoltaics, serves as an excellent benchmark for complex chemistry simulations. Its electronic structure calculation involves measuring Hamiltonians comprising thousands of Pauli strings, even for modest active spaces, making precision a formidable challenge [55]. The requirement for chemical precision (1.6 × 10â»Â³ Hartree) in energy estimation sets a high bar for quantum algorithm performance amidst noise [55].
Experimental Protocols for Noisy Quantum Simulations
High-Precision Energy Estimation Protocol
Achieving chemical precision on NISQ devices requires a multi-faceted strategy that addresses both statistical and systematic errors. The following protocol, validated on the BODIPY molecule, outlines this process [55]:
- State Preparation: Initialize the system into the Hartree-Fock state. This separable state is chosen to isolate measurement errors from two-qubit gate errors, providing a clear baseline for analyzing measurement noise [55].
- Informationally Complete (IC) Measurements: Implement a set of measurements that fully characterizes the quantum state. This allows for the estimation of multiple observables from the same dataset and provides a foundation for advanced error mitigation [55].
- Locally Biased Random Measurements: To reduce the shot overhead (number of measurements), prioritize measurement settings that have a larger impact on the final energy estimation. This strategy, known as Hamiltonian-inspired locally biased classical shadows, optimizes the use of limited quantum resources [55].
- Quantum Detector Tomography (QDT): To mitigate readout errors, perform parallel QDT to characterize the noisy measurement apparatus. The resulting model is used to build an unbiased estimator for the molecular energy, significantly reducing estimation bias [55].
- Blended Scheduling: Execute the circuits for Hamiltonian measurement and QDT in an interleaved, or "blended," manner. This technique averages out temporal fluctuations in device noise, ensuring that all parts of the experiment are subject to the same average noise conditions, which is critical for homogeneous energy gap estimations (e.g., Sâ‚€, Sâ‚, Tâ‚) [55].
This integrated approach has demonstrated a reduction in measurement errors by an order of magnitude, from 1-5% down to 0.16%, on an IBM Eagle r3 quantum processor [55].
Noise-Resilient State Preparation via Imaginary Time Evolution
Imaginary Time Evolution (ITE) is a non-unitary method for preparing ground states. Its robustness under noise can be analyzed using a Trotterized model applied to the Transverse-Field Ising Hamiltonian [56]:
H = -J ∑σz_i σz_{i+1} + g ∑σx_i
The following diagram illustrates the conceptual workflow for analyzing a noisy ITE process:
The process involves iteratively applying a Trotter step of ITE, followed by a noise channel, until the state converges to a noisy steady state. Research reveals that the ordered phase and associated phase transition of the Ising model persist in the presence of noise, provided the noise does not explicitly break the symmetry protecting the phase [56]. This demonstrates an inherent resilience in certain state preparation protocols.
The Scientist's Toolkit: Research Reagent Solutions
Successful experimentation in noisy quantum environments relies on a suite of computational "reagents." The following table details essential tools and their functions for molecular ground state modeling.
Table 2: Essential Research Reagents for Noisy Quantum Chemistry Simulations
| Research Reagent | Function & Purpose | Example Use-Case |
|---|---|---|
| OMol25 Dataset | Massive dataset of 100M+ DFT calculations for training ML potentials and benchmarking; provides high-accuracy reference data [57] [53]. | Training neural network potentials (NNPs) for fast, DFT-accurate energy predictions on large systems. |
| Neural Network Potentials (NNPs) | Machine-learning models trained on OMol25; predict energies/forces with DFT accuracy but ~10,000x faster [57] [53]. | Running long-time molecular dynamics simulations on large biomolecules or electrolytes. |
| Universal Model for Atoms (UMA) | A pre-trained, universal NNP unifying OMol25 with other datasets; functions "out-of-the-box" for diverse applications [57]. | Rapid property prediction and geometry optimization without task-specific model training. |
| Variational Quantum Eigensolver (VQE) | A hybrid quantum-classical algorithm to find molecular ground states by minimizing energy with respect to a parameterized quantum circuit [54]. | Estimating ground state energies of small molecules like BODIPY on near-term quantum hardware. |
| Compact Heuristic Circuit (CHC) Ansatz | A parameterized quantum circuit designed for the VQE that reduces circuit complexity/depth without sacrificing accuracy [54]. | Mitigating noise accumulation in VQE for molecular vibrational energy calculations. |
| Error Mitigation Framework (APGEM-ZNE-PEC) | A hybrid framework integrating Adaptive Policy-Guided Error Mitigation, Zero-Noise Extrapolation, and Probabilistic Error Cancellation [30]. | Stabilizing Quantum Reinforcement Learning algorithms against depolarizing and amplitude damping noise. |
| FPS-ZM1 | FPS-ZM1|RAGE Inhibitor|For Research Use | |
| FR-188582 | FR-188582, CAS:189699-82-9, MF:C16H13ClN2O2S, MW:332.8 g/mol | Chemical Reagent |
Integrated Error Mitigation and Workflow
No single technique is sufficient to overcome the limitations of NISQ devices. A robust workflow integrates multiple mitigation strategies, as shown in the following comprehensive diagram.
This integrated workflow is critical for countering phenomena like Noise-Induced Barren Plateaus (NIBPs), where noise causes the gradients of VQA cost functions to vanish exponentially, rendering optimization impossible [5]. While unital noise (e.g., depolarizing) always induces NIBPs, some non-unital noise (e.g., amplitude damping) may be less detrimental, highlighting the need for noise-aware strategy selection [5].
Modeling molecular ground states under realistic noise conditions remains a significant challenge, yet substantial progress is being made. The synergistic combination of advanced datasets like OMol25, noise-resilient state preparation protocols, and integrated error mitigation frameworks provides a viable path forward. The experimental protocols and toolkit detailed in this study offer researchers a blueprint for achieving chemically precise results on current quantum hardware. As quantum processors improve and error mitigation strategies mature, the quantum-classical approach to computational chemistry will increasingly enable the exploration of complex molecular systems that are currently beyond reach, accelerating discovery in drug development and materials science.
The accurate prediction of protein-ligand binding affinity is a critical challenge in computational drug discovery. While classical computational methods have made significant advances, they often face limitations in accuracy or require prohibitive computational resources for large-scale virtual screening. The emergence of quantum computing, particularly in the Noisy Intermediate-Scale Quantum (NISQ) era, offers promising avenues to overcome these limitations. This technical guide explores the integration of quantum machine learning (QML) and hybrid quantum-classical algorithms for predicting protein-ligand binding energies, with particular focus on their resilience to realistic quantum noise models including depolarizing and amplitude damping channels. The inherent noise in current quantum devices presents significant challenges for quantum chemistry simulations, yet recent research demonstrates that certain algorithmic approaches can not only mitigate these effects but potentially leverage them for more efficient computations in specific scenarios.
Quantum Approaches to Binding Affinity Prediction
Hybrid Quantum Neural Networks for Binding Affinity
Hybrid Quantum Neural Networks (HQNNs) represent a promising architecture for NISQ-era applications in drug discovery. These models integrate classical deep learning frameworks with parameterized quantum circuits, replacing specific classical layers with quantum components. For protein-ligand binding affinity prediction, the HQDeepDTAF framework has been developed, inspired by the classical DeepDTAF architecture but substituting classical neural networks with hybrid quantum models [58].
The HQDeepDTAF architecture consists of three separate modules processing different molecular representations: the entire protein module, the local pocket module, and the ligand SMILES (Simplified Molecular Input Line Entry System) module. A key innovation in this approach is the implementation of data re-uploading models within the hybrid quantum framework, which addresses limitations in the expressivity of pure Quantum Neural Networks (QNNs). To maintain feasibility on NISQ devices, the researchers introduced a hybrid embedding scheme that reduces required qubit counts compared to standard encoding methods, utilizing classical regression networks for the final prediction task [58].
Numerical results indicate that HQNNs achieve comparable or superior performance to classical neural networks while offering improved parameter efficiency. This reduction in parameters while maintaining performance is particularly valuable for drug discovery applications where training computational models on large molecular datasets can be prohibitively expensive [58].
Quantum Convolutional Algorithms
An alternative QML approach specifically designed for structure-based virtual screening is the Quantum Convolutional Algorithm for Drug Discovery (QCADD). This method encodes molecular information into quantum states using nine qubits that represent atom types and spatial coordinates, processing this data through a Quantum Convolutional Neural Network (QCNN) with layers of single-qubit rotations and entanglement layers [59].
The QCADD architecture is carefully balanced to maximize expressive power while remaining feasible on current NISQ devices. To enhance scalability, the researchers developed a method for parallel estimation that processes multiple protein-ligand complexes simultaneously using ancillary qubits. Results demonstrate that circuits with five to six quantum layers consistently yield the most accurate predictions, achieving a remarkable root mean square deviation (RMSD) of 2.37 kcal/mol for binding energy predictions alongside a Pearson correlation of 0.650 with experimental data [59].
Notably, the model maintains relative ranking of ligand affinities even under noisy conditions, a critical feature for virtual screening where prioritization of candidate compounds is more important than absolute binding energy values [59].
Performance and Noise Resilience
Quantitative Performance Metrics
Table 1: Performance Metrics of Quantum Approaches for Binding Affinity Prediction
| Algorithm | Prediction Accuracy | Noise Resilience | Key Advantages |
|---|---|---|---|
| HQDeepDTAF (Hybrid Quantum) | Comparable or superior to classical NN | Maintains performance with parameter efficiency | Reduced parameters, feasible on NISQ devices |
| QCADD (Quantum Convolutional) | RMSD: 2.37 kcal/mol, Pearson: 0.650 | Maintains ligand ranking under noise | Parallel estimation for scalability |
| Quanvolutional NN (Image-based) | ~30% higher accuracy than QCNN in validation | Greater robustness across multiple noise channels | Flexible circuit architecture |
Table 2: Impact of Different Noise Channels on Quantum Models
| Noise Channel | Impact on Quantum Models | Notable Characteristics |
|---|---|---|
| Depolarizing Noise | Universally suppresses magic, cannot generate it | Effectively simulates isotropic experimental noise |
| Amplitude Damping | Can generate or enhance magic depending on input state | Non-unital channel representing energy dissipation |
| Phase Damping | Affects coherence without energy loss | Models pure dephasing processes in qubits |
| Bit Flip | Alters computational basis states | Can be mitigated through error correction |
| Phase Flip | Affects phase information in qubits | Equivalent to Z-error in quantum error correction |
Comparative Analysis of Noise Resilience
Recent comprehensive studies have evaluated the robustness of various HQNN architectures against different quantum noise channels. In comparative analyses of Quantum Convolution Neural Networks (QCNN), Quanvolutional Neural Networks (QuanNN), and Quantum Transfer Learning (QTL), researchers found that QuanNN generally demonstrates greater robustness across multiple quantum noise channels, consistently outperforming other models [21].
The performance variation between different HQNN architectures under identical experimental settings can be significant - with QuanNN outperforming QCNN by approximately 30% in validation accuracy under certain conditions [21]. This highlights the importance of architectural selection for specific noise environments in NISQ devices.
Interestingly, certain types of noise can be leveraged rather than merely mitigated. Research on nonstabilizerness (magic) in quantum circuits has revealed that amplitude damping - a nonunital channel representing energy dissipation - can actually generate or enhance magic depending on the input state and noise strength, whereas depolarizing noise universally suppresses magic and cannot generate it [20]. This suggests that for specific computational tasks, the noise characteristics of quantum hardware could be matched to algorithmic requirements.
Experimental Protocols and Methodologies
Implementation of HQDeepDTAF
The experimental protocol for implementing and evaluating the HQDeepDTAF framework involves several critical steps:
Data Preparation and Feature Extraction: Molecular structures from the PDBbind database are processed to extract features for the three model inputs: entire protein sequences, local binding pocket information, and ligand SMILES strings [58].
Hybrid Embedding Scheme: Classical molecular features are encoded into quantum states using a hybrid embedding approach designed to reduce qubit requirements. This involves mapping classical features into quantum state space while maintaining constant circuit depth to accommodate NISQ device limitations [58].
Quantum Circuit Configuration: The quantum component employs data re-uploading circuits with carefully selected numbers of qubits and layers based on two key metrics: expressibility and entangling capability. This configuration is optimized through numerical simulations before implementation on quantum hardware [58].
Noise Simulation and Mitigation: The model is evaluated under simulated noise conditions matching real NISQ devices, including depolarizing and amplitude damping channels. Noise resilience techniques are incorporated based on the specific error characteristics [58].
Performance Validation: The trained model is validated on test datasets comparing performance metrics (typically mean absolute error and correlation coefficients) against classical benchmarks and experimental data where available [58].
QCADD Implementation Protocol
The experimental methodology for the Quantum Convolutional Algorithm for Drug Discovery involves:
Molecular Encoding: Protein-ligand complexes are represented as 3D structures with atom types and spatial coordinates encoded into nine qubits using angle embedding techniques [59].
Quantum Circuit Design: A parameterized quantum circuit is constructed with multiple layers of single-qubit rotations (RY, RZ gates) and entanglement layers (CNOT gates) arranged in a specific pattern to balance expressivity and trainability [59].
Resource Optimization: Circuit depth is optimized to 5-6 quantum layers based on systematic testing showing optimal performance at this depth. Parallel estimation techniques using ancillary qubits are implemented for processing multiple complexes simultaneously [59].
Noise Resilience Testing: The model is evaluated under conditions of limited sampling (shot noise) and simulated hardware noise including depolarizing and amplitude damping channels. Performance is assessed both on absolute binding energy accuracy and preservation of ligand ranking [59].
Classical Integration: Quantum predictions are processed through classical post-processing layers to generate final binding affinity estimates, with comparative analysis against classical molecular docking and machine learning approaches [59].
Diagram 1: QCADD Experimental Workflow. The protocol begins with molecular structure encoding, progresses through quantum computation with integrated noise testing, and concludes with classical post-processing for final prediction.
Table 3: Essential Research Resources for Quantum-Enhanced Binding Affinity Calculations
| Resource Category | Specific Tools/Platforms | Function in Research |
|---|---|---|
| Quantum Simulators | Qiskit, Cirq, Pennylane | Simulation of quantum algorithms with configurable noise models before hardware deployment |
| Molecular Databases | PDBbind Database [58] | Provides curated protein-ligand complexes with experimental binding affinity data for training and validation |
| Classical Force Fields | ARROW Force Field [60] | Multipolar polarizable physics-based model for reference calculations and hybrid approaches |
| Enhanced Sampling | HREX with Conformation Reservoir [60] | Hamiltonian Replica Exchange method coupled with nonequilibrium MD for improved conformational sampling |
| Error Mitigation | Pauli Saving [61], Ansatz-based Techniques | Reduces measurement costs and mitigates hardware errors in subspace methods |
| Hardware Platforms | IBM Quantum (ibm-cleveland) [62] | NISQ devices for experimental validation of quantum algorithms under realistic noise conditions |
Noise Resilience Mechanisms and Theoretical Framework
Understanding Noise Effects in Quantum Chemistry Calculations
The performance of quantum algorithms for chemistry simulations on NISQ devices is fundamentally constrained by various noise channels. Research has revealed that different types of quantum noise affect algorithms in distinct ways:
Depolarizing noise represents an isotropic error model where qubits randomly transition to completely mixed states with some probability. This type of noise universally suppresses nonstabilizerness (magic) in quantum circuits and cannot generate it, making it particularly detrimental for algorithms relying on quantum magic for computational advantage [20].
Amplitude damping models energy dissipation from qubits to their environment, representing a nonunital channel that can actually generate or enhance magic depending on the input state and noise strength. This counterintuitive effect suggests that certain noise types could potentially be leveraged rather than merely mitigated in specific computational contexts [20].
Measurement noise including shot noise fundamentally limits the precision of quantum measurements, requiring sophisticated techniques like Pauli saving to reduce measurement costs, particularly for quantum subspace methods and linear response techniques [61].
Effective Lindbladian Models for Noisy Quantum Simulations
Recent theoretical advances propose reframing the impact of noise in quantum simulations through the lens of effective Lindbladians. Rather than interpreting noise as purely detrimental, this approach recognizes that when simulating physical systems on noisy quantum hardware, the combined effect of Trotterized time evolution and device noise can be described as simulating a different, effectively open quantum system [63].
This noisy algorithm model reinterprets the effects of hardware noise as a shift to the dynamics of the original system being simulated, described through static Lindblad noise terms that act in addition to the original unitary dynamics. The form of these effective noise terms depends not only on the underlying hardware noise processes but also on the original unitary dynamics and the specific quantum algorithm chosen for simulation [63].
This framework is particularly relevant for protein-ligand binding simulations, as biological systems naturally experience environmental interactions that might be effectively captured through these emergent noise models in quantum simulations.
Diagram 2: Divergent Impact of Noise Channels on Quantum Resources. Different noise types have opposing effects on nonstabilizerness ("magic"), a key resource for quantum advantage.
Future Directions and Implementation Considerations
The integration of quantum computing for protein-ligand binding affinity prediction is rapidly evolving, with several promising directions for further research and development. The current state of research suggests that hybrid quantum-classical approaches will dominate practical applications in the near term, leveraging classical resources for preprocessing and postprocessing while utilizing quantum circuits for specific computationally intensive subtasks.
Key considerations for implementing these approaches include:
Resource Allocation: Balancing quantum circuit depth and width against available qubit coherence times and gate fidelities on target hardware platforms.
Algorithm Selection: Matching specific quantum algorithms (QuanNN, QCNN, VQE variants) to particular protein-ligand systems based on molecular complexity and available training data.
Error Mitigation Strategy: Implementing application-specific error mitigation techniques rather than generic approaches, potentially leveraging noise characteristics rather than simply combating them.
Validation Protocols: Establishing robust benchmarking methodologies that account for both classical and quantum sources of error in binding affinity predictions.
As quantum hardware continues to improve with increasing qubit counts, enhanced coherence times, and lower gate errors, the balance between classical and quantum resources in these hybrid frameworks will inevitably shift. However, the insights gained from current noise-resilient algorithm development will continue to inform the design of future quantum computing applications in drug discovery and molecular simulation.
Hybrid Quantum-Classical Workflows for Practical Pharmaceutical Applications
The integration of hybrid quantum-classical computing into pharmaceutical research represents a paradigm shift with the potential to radically accelerate drug discovery and development. By leveraging the complementary strengths of quantum and classical systems, researchers can now tackle problems that have long been intractable for classical computing alone, particularly in molecular simulation and optimization. This whitepaper provides a technical examination of these workflows, with specific attention to the impact of quantum noise models—including depolarizing and amplitude damping—on the simulation of chemical systems. We detail experimental protocols, error mitigation strategies, and provide a practical toolkit for researchers embarking on this transformative path. Framed within the context of a broader thesis on noise resilience, this guide serves as a roadmap for the integration of quantum-classical methods into the pharmaceutical development pipeline, offering a realistic perspective on both current capabilities and future horizons.
The pharmaceutical industry faces a persistent challenge of declining R&D productivity, driven by the high failure rates of drug candidates during development, the shift toward more complex biologics, and the focus on poorly understood diseases [64]. Traditional computational methods, including AI and molecular dynamics simulations, are often hampered by their inability to accurately model quantum-level interactions, such as electron correlations, which are fundamental to predicting molecular behavior [64] [65]. Hybrid quantum-classical workflows emerge as a powerful solution to this impasse. These workflows strategically partition computational tasks, using classical computers for data management and well-understood simulations, while delegating specific, computationally prohibitive sub-problems—most notably, the electronic structure calculation of molecular systems—to quantum co-processors [66].
The value at stake is substantial; quantum computing in life sciences is estimated to create $200 billion to $500 billion in value by 2035 [64]. This value stems from quantum computing's unique capability to perform first-principles calculations based on the laws of quantum physics, enabling highly accurate, predictive, in silico research. This is not merely an incremental improvement but a fundamental change that could transform the entire pharmaceutical value chain, from initial discovery to patient delivery [64]. The core of this transformation lies in the ability to simulate molecular interactions from scratch, without relying on existing experimental data, thereby significantly reducing the need for lengthy and costly wet-lab experiments [64].
Workflow Architecture and Core Components
A hybrid workflow is an integrated pipeline where quantum and classical resources operate in concert. The typical architecture for a pharmaceutical application involves a recursive cycle of problem preparation, quantum execution, and classical post-processing.
The Hybrid Workflow Loop
The following diagram illustrates the continuous, interactive process of a hybrid quantum-classical workflow.
This workflow is most commonly implemented through Variational Quantum Algorithms (VQAs), such as the Variational Quantum Eigensolver (VQE). In the context of drug discovery, the "cost function" is often the electronic energy of a molecule, which the classical optimizer seeks to minimize by iteratively updating the parameters of the quantum circuit [30] [5].
Key Application Areas in Drug Discovery
Hybrid workflows are being applied to several critical problems in pharmaceuticals:
- Protein-Ligand Binding and Hydration: Accurately predicting how a small molecule (ligand) binds to a protein target is fundamental. A key advancement involves analyzing protein hydration, where water molecules mediate binding. Collaborations, such as that between Pasqal and Qubit Pharmaceuticals, have developed hybrid quantum-classical approaches to precisely place water molecules within protein pockets, a task that is computationally demanding for purely classical methods [67].
- Electronic Structure Simulations: Understanding the electronic structure of molecules, especially complex systems like metalloenzymes which are critical for drug metabolism, is a primary application. Companies like Boehringer Ingelheim are collaborating with quantum hardware firms to explore these calculations, which are vital for predicting drug interactions and efficacy [64].
- Molecular Docking and Off-Target Effect Prediction: Quantum computing can provide more reliable predictions of binding strength (docking) and can simulate reverse docking to identify potential side effects and toxicity early in the development process, thereby reducing the risk of late-stage failures [64].
Quantitative Benchmarks and Industry Progress
The field is transitioning from theoretical promise to tangible benchmarks. The table below summarizes key quantitative milestones demonstrating the progress and potential of hybrid quantum-classical applications.
Table 1: Key Performance Benchmarks in Hybrid Quantum-Classical Computing
| Application Area | Reported Performance | System Used | Significance |
|---|---|---|---|
| Medical Device Simulation | Outperformed classical HPC by 12% [68] | IonQ's 36-qubit computer | One of the first documented cases of practical quantum advantage in a real-world application. |
| Algorithm Execution Speed | 13,000x faster than classical supercomputers [68] | Google's Willow quantum chip | Demonstrates verifiable quantum advantage for specific algorithms. |
| Quantum Error Correction | Error rates reduced to 0.000015% per operation [68] | Various (QuEra, Microsoft) | Critical for achieving fault-tolerant quantum computation, enabling more complex simulations. |
| Development Efficiency | 70%+ reduction in dev time; 100-1000x cost reduction [69] | Qoro's software stack | Shows the impact of software and middleware in making quantum workflows practical and efficient. |
| Generative Model Quality | FID score of ~2.28 on FFHQ dataset [66] | ORCA's hybrid GAN (simulated) | A record for a hybrid quantum-classical generative model, indicating high-quality data generation. |
These benchmarks are supported by a robust investment landscape. The global quantum computing market is projected to grow at a compound annual growth rate (CAGR) of 32.7% to 41.8%, potentially reaching $20.2 billion by 2030 [68]. This financial momentum underscores the confidence in the technology's future impact.
Experimental Protocols for Molecular Simulation
This section provides a detailed methodology for conducting a molecular simulation, such as calculating the ground state energy of a drug candidate, using a hybrid VQE approach under noisy conditions.
Protocol: VQE with Integrated Noise Modeling
Objective: To compute the ground state energy of a target molecule (e.g., a small hydrocarbon or a fragment of a protein) while characterizing and mitigating the impact of quantum noise.
Materials & Prerequisites:
- Classical Computing Resources: A high-performance computing (HPC) node equipped with a quantum circuit simulator (e.g., Qiskit Aer) and a classical optimizer (e.g., COBYLA, SPSA).
- Problem Definition: The second-quantized molecular Hamiltonian of the target system, derived through a classical computational chemistry package (e.g., PySCF).
- Quantum Ansatz: A parameterized quantum circuit (ansatz), such as the Unitary Coupled Cluster (UCC) ansatz or a hardware-efficient ansatz.
Procedure:
- Hamiltonian Encoding: Map the fermionic molecular Hamiltonian to a qubit Hamiltonian using a transformation such as Jordan-Wigner or Bravyi-Kitaev.
- Ansatz Initialization: Select and initialize the parameterized quantum circuit. The number of qubits will correspond to the number of spin orbitals in the problem.
- Noise Model Configuration: Define the noise model to be investigated. This involves setting parameters for:
- Depolarizing Noise: Characterized by a probability
p_depolthat a single-qubit or two-qubit gate is replaced by a completely mixed state. - Amplitude Damping: Characterized by a relaxation time
T1, which models energy dissipation from the qubit to its environment.
- Depolarizing Noise: Characterized by a probability
- Quantum Circuit Execution: For each set of parameters
θfrom the classical optimizer: a. Prepare the initial state (usually |0>^⊗n). b. Apply the parameterized ansatz circuit. c. Measure the expectation values of the terms in the qubit Hamiltonian. d. This step is repeated multiple times (shots) to build statistical confidence. - Classical Computation of Cost Function: On the classical processor, compute the total energy expectation value
E(θ)by summing the measured expectation values of the Hamiltonian terms. - Classical Optimization: The classical optimizer analyzes
E(θ)and proposes a new set of parametersθ_newto lower the energy. - Iteration and Convergence: Steps 4-6 are repeated until the energy
E(θ)converges to a minimum, within a predefined tolerance, or for a maximum number of iterations.
Data Analysis and Noise Impact Assessment
- Convergence Trajectory: Plot the energy
E(θ)against the iteration number. Compare the convergence behavior under different noise models (depolarizing vs. amplitude damping) and against a noiseless simulation. - Final Energy Accuracy: Calculate the error between the final computed energy and the exact classical result (if available) or a high-accuracy benchmark. This quantifies the overall impact of noise on the solution's quality.
- Parameter Stability: Monitor the variation in the optimal parameters
θunder different noise conditions. Large shifts can indicate that the noise is pulling the solution towards a noise-induced optimum.
Quantum Noise Models and Mitigation Strategies
The performance of hybrid workflows on current Noisy Intermediate-Scale Quantum (NISQ) devices is fundamentally constrained by quantum noise. A detailed understanding of noise models and their mitigation is essential for obtaining meaningful results.
Relevant Noise Models for Chemistry Simulations
- Depolarizing Noise: A unital noise model that, with probability
p, replaces the quantum state with the maximally mixed state, effectively introducing white noise. It is a common model for gate imperfections. Research has shown that depolarizing noise can introduce significant randomness, leading to severe performance degradation and is a key contributor to Noise-Induced Barren Plateaus (NIBPs), where the cost function gradient vanishes exponentially with system size [30] [5]. - Amplitude Damping: A non-unital noise model that describes the energy loss of a qubit to its environment, characterized by the
T1relaxation time. This model is physically grounded and particularly relevant for simulating molecular relaxation processes. Recent studies classify it as Hilbert-Schmidt (HS)-contractive and show that while it still degrades performance, it may not always lead to NIBPs, unlike depolarizing noise. Instead, it can cause Noise-Induced Limit Sets (NILS), where the cost function converges to a range of values rather than a single minimum [5]. - Other Models: Phase damping/dephasing (characterized by
T2time) and measurement noise also play a role, though measurement noise typically has a milder effect as it primarily impacts the readout stage [30].
A Framework for Error Mitigation
A robust error mitigation strategy is multi-layered. The following diagram outlines a hybrid framework that combines techniques at both the circuit and algorithmic levels.
Table 2: Error Mitigation Techniques for Pharmaceutical Simulations
| Technique | Mechanism | Advantages | Challenges |
|---|---|---|---|
| Zero-Noise Extrapolation (ZNE) | Intentionally increases noise (e.g., by stretching gates), then extrapolates back to the zero-noise result [30]. | Conceptually simple, no prior device calibration needed. | Can amplify errors; relies on accurate noise models for extrapolation. |
| Probabilistic Error Cancellation (PEC) | Uses a pre-calibrated noise model to construct "anti-noise" gates, which are applied stochastically to cancel errors [30]. | Can, in theory, completely cancel known errors. | Requires extensive and frequent device calibration; introduces sampling overhead. |
| Adaptive Policy-Guided Error Mitigation (APGEM) | An algorithmic-level technique that uses reinforcement learning to adapt the optimization policy based on reward trends, stabilizing learning despite noise fluctuations [30]. | Increases robustness and learning stability; can be combined with circuit-level techniques. | Adds computational complexity to the classical optimization loop. |
Recent research demonstrates that a hybrid mitigation framework synergistically integrating APGEM, ZNE, and PEC can yield marked improvements in convergence stability, solution quality, and informational coherence under realistic noise conditions [30].
The Scientist's Toolkit: Research Reagent Solutions
Transitioning from theory to practice requires a suite of software and hardware "reagents." The following table details essential components for building and executing hybrid quantum-classical workflows.
Table 3: Essential Resources for Hybrid Workflow Implementation
| Tool Category | Example Platforms & Tools | Primary Function |
|---|---|---|
| Quantum Hardware Access | IBM Quantum, IonQ, Pasqal's Orion, ORCA PT-Series [64] [67] [66] | Provides access to physical quantum processing units (QPUs) for algorithm execution. |
| Hybrid Software Platforms | NVIDIA CUDA-Q, Qoro Stack, ORCA SDK [66] [69] | Software development platforms that facilitate the integration and orchestration of quantum and classical computing resources. |
| Quantum Simulators | Qiskit AerSimulator [30] [70] | Simulates quantum circuits on classical HPC systems, essential for algorithm development and testing under controlled (noisy or noiseless) conditions. |
| Error Mitigation Libraries | Built into frameworks like Qiskit; custom implementations of ZNE, PEC, etc. [30] [70] | Provide pre-built functions to apply techniques like ZNE and PEC to quantum circuits before and after execution. |
| Molecular Modeling & Chemistry | qBraid's Quanta-Bind, Classical computational chemistry suites (e.g., PySCF) [71] [65] | Tools for generating molecular Hamiltonians, preparing chemical problems for quantum simulation, and analyzing results. |
| ICG-001 | ICG-001, CAS:780757-88-2, MF:C33H32N4O4, MW:548.6 g/mol | Chemical Reagent |
| AG1557 | AG1557, CAS:1161002-05-6, MF:C19H16BrNO2, MW:370.2 g/mol | Chemical Reagent |
Hybrid quantum-classical workflows are rapidly evolving from experimental curiosities into essential tools for pharmaceutical R&D. The core challenge of noise is being met with increasingly sophisticated mitigation strategies, making it possible to run meaningful chemistry simulations on today's NISQ devices. The demonstrated capabilities in simulating protein-ligand binding, electronic structures, and molecular properties underscore a clear path toward quantum utility in drug discovery.
The future trajectory points toward the tighter integration of these workflows into the broader computational landscape. This includes their role in generating high-quality data for training AI models [64], the emergence of quantum machine learning (QML) for trial optimization [64], and the use of AI to design better quantum hardware and circuits, as seen with ORCA's RS-GPT algorithm [66]. For researchers and drug development professionals, the imperative is to build strategic alliances, invest in multidisciplinary talent, and begin actively experimenting with these workflows to develop the in-house expertise required to leverage this transformative technology fully [64]. Companies that engage early will be best positioned to accelerate their research, reduce costs, and ultimately deliver life-changing therapies to patients more quickly.
Advanced Error Mitigation and Optimization Strategies for Noise-Resilient Chemical Predictions
Zero-Noise Extrapolation (ZNE) Techniques for Enhancing Molecular Energy Estimates
Accurately calculating molecular energies is a cornerstone of computational chemistry and drug development, with the potential to revolutionize material design and therapeutic discovery. On noisy intermediate-scale quantum (NISQ) devices, quantum noise remains a fundamental barrier to achieving the precision required for practical applications. Zero-noise extrapolation (ZNE) has emerged as a leading error mitigation strategy that does not require additional qubits, making it particularly suitable for the constraints of current quantum hardware. This technique operates on a fundamentally simple principle: systematically amplifying the inherent noise in a quantum circuit, measuring the observable of interest at these elevated noise levels, and then extrapolating back to estimate the expected value at the zero-noise limit [72].
Within the specific context of molecular energy calculations—such as determining the ground state energy of molecules using the Variational Quantum Eigensolver (VQE) algorithm—the deleterious effects of noise can lead to significant overestimation of energies and failure to locate correct molecular geometries [73]. This technical guide provides an in-depth examination of ZNE techniques, with a focused analysis on their application to molecular energy estimates, benchmarking under realistic noise models, and integration into robust quantum chemistry simulation workflows.
Foundations of Zero-Noise Extrapolation
Core Principles and Mathematical Framework
The fundamental premise of ZNE is that the relationship between an observed expectation value and the underlying noise strength can be modeled and extrapolated. For a target observable, the noisy expectation value ( E(\lambda) ) is measured at multiple, intentionally scaled noise strengths ( \lambda \geq 1 ), where ( \lambda = 1 ) represents the base level of hardware noise. A functional form for ( E(\lambda) ) is assumed, and its parameters are fitted to the measured data. The estimate for the noiseless expectation value ( E_0 ) is then obtained by evaluating the fitted function at ( \lambda = 0 ) [72].
The most common analytical approach, Richardson extrapolation, relies on polynomial interpolation. Despite its widespread use, it faces challenges related to approximation errors from the non-polynomial behavior of physical noise channels, overfitting, and the exponential amplification of measurement noise as the extrapolation range increases [72].
A general noise model for quantum circuits can be formulated using perturbative expansion. Consider a circuit where the imperfect execution of a two-qubit gate ( g ) is described by ( G{\mathcal{N}} = \mathcal{N}g \circ g ), with the noise channel ( \mathcal{N}g ) modeled as: [ \mathcal{N}g = \mathcal{I} + \sum{i=1}^{d} qg^i \mathcal{E}i ] Here, ( \mathcal{I} ) is the identity channel, ( \mathcal{E}i ) are gate-independent noise operators, and ( qg^i ) are the associated error rates. The resulting noisy output state ( \rho ) and the expectation value ( E = \operatorname{Tr}(\rho H) ) for observable ( H ) can be expressed as a power series in the error rates ( qg^i ): [ E = E0 + \sum{i=1}^{d} \sum{g \in T} qg^i Eg^i + O(q^2) ] where ( E0 ) is the noiseless expectation value and ( E_g^i ) are the first-order error coefficients [74]. ZNE aims to cancel out these leading-order error terms through its extrapolation procedure.
Promising ZNE Variants for Chemistry Applications
Recent research has moved beyond basic Richardson extrapolation to develop more powerful and hardware-efficient ZNE variants.
Cyclic Layout Permutations ZNE (CLP-ZNE): This method leverages the non-uniformity of gate errors across a quantum processor. Instead of physically scaling gate noise, the same logical circuit is executed over multiple qubit layouts that cyclically permute the circuit's connectivity path. For an ( n )-qubit circuit with linear connectivity, only ( O(n) ) different layouts are needed to construct an extrapolation to the zero-noise limit. This approach provably mitigates both unital and non-unital noise without adding any extra gates, making it highly suitable for the deep circuits often encountered in quantum chemistry [74].
Noise-Robust Estimation (NRE): A novel noise-agnostic framework that addresses the model mismatch problem inherent in standard ZNE. NRE introduces a two-step post-processing technique. First, it constructs a baseline error-mitigated estimation. Then, it discovers and exploits a statistical correlation between the residual bias of this baseline and a measurable quantity called the normalized dispersion. By extrapolating to the zero-dispersion limit, NRE can suppress residual bias without requiring an explicit noise model, achieving near bias-free estimation of energies for molecules like H4, even at high circuit depths [75].
ZNE for Molecular Energy Calculations: Protocols and Workflows
General VQE Workflow with Integrated ZNE
The following diagram illustrates the standard workflow for a Variational Quantum Eigensolver that incorporates ZNE for mitigating errors in molecular energy calculations.
Figure 1: VQE with ZNE Workflow. This flowchart outlines the hybrid quantum-classical loop for calculating molecular ground state energy with integrated Zero-Noise Extrapolation. The key ZNE-specific steps of noise scaling and extrapolation are embedded within each energy evaluation cycle.
The process begins by defining the molecule of interest and generating its electronic Hamiltonian. A parameterized quantum circuit (ansatz), such as the Unitary Vibrational Coupled Cluster (UVCC) or the more compact Compact Heuristic Circuit (CHC), is then prepared [54]. Before execution, the circuit's noise is scaled to several predefined levels ( \lambda_i ) using techniques like gate folding or pulse stretching [72]. Each scaled circuit is executed, and the energy expectation value ( E(\lambda) ) is measured. These noisy energy points are used to extrapolate the zero-noise estimate ( E(0) ). A classical optimizer then uses this mitigated energy to update the circuit parameters, and the loop repeats until convergence is reached.
Protocol for Molecular Geometry Optimization
Determining a molecule's most stable structure is a key application. The following protocol, demonstrated for the trihydrogen cation (H₃âº) on Amazon Braket using IQM's Garnet device calibration data, shows how to integrate ZNE [73].
- Problem Encoding: Encode the H₃⺠molecule (2 electrons, 3 hydrogen atoms) onto 6 qubits. The objective is the joint minimization of the energy ( E(\theta, x) = \langle \psi(\theta) | H(x) | \psi(\theta) \rangle ) with respect to both the circuit parameters ( \theta ) and the nuclear coordinates ( x ).
- Noise Model Construction: Build a noise model from hardware calibration data. Import predefined noise channels (
AmplitudeDamping,Depolarizing,PhaseDamping,TwoQubitDepolarizing) and scale their probabilities using the latest device parameters (Tâ‚, Tâ‚‚, gate fidelities, readout errors) [70] [73]. - Circuit Execution with ZNE: For each candidate molecular geometry ( x ) and parameters ( \theta ) during the optimization:
- Scale the base noise model to multiple factors (e.g., ( \lambda = [1, 2, 3] )).
- Run the parameterized circuit for each ( \lambda ) on a simulator emulating the constructed noise model (or directly on a QPU).
- Record the measured energies ( E(\lambda) ).
- Extrapolation: Fit a polynomial (e.g., linear or quadratic) to the data points ( (\lambda, E(\lambda)) ) and evaluate it at ( \lambda = 0 ) to obtain the mitigated energy estimate for the classical optimizer.
- Validation: The successful protocol should converge to the known equilibrium geometry of H₃âº: an equilateral triangle with a bond length of 0.985 Ã… and a ground state energy of -1.274 Ha [73].
Performance Analysis and Benchmarking
Quantitative Performance Under Different Noise Models
The efficacy of ZNE and its variants varies significantly depending on the underlying noise channel and the system being simulated. The table below summarizes key performance metrics from recent studies.
Table 1: Performance Benchmarks of ZNE and Advanced Variants
| Method / Model | System / Circuit | Key Performance Metric | Result |
|---|---|---|---|
| CLP-ZNE [74] | 12-qubit Sherrington-Kirkpatrick (SK) model | Error reduction factor vs. unmitigated | 8x to 13x reduction (IBM Torino noise model) |
| CLP-ZNE [74] | Transverse Field Ising Model (TFIM) | Error suppression under non-unital noise | Significant suppression under strong $T1/T2$ relaxation |
| NRE [75] | H4 molecule (quantum chemistry) | Accuracy in ground-state energy recovery | Restored energy close to chemical accuracy despite high circuit depth |
| NRE [75] | Transverse Field Ising Model (TFIM) | Remaining bias error vs. other QEM methods | Outperformed ZNE, CDR, vnCDR; bias reduced by up to 2 orders of magnitude |
| Standard ZNE [73] | H₃⺠geometry optimization (VQE) | Ability to find correct equilibrium geometry | Enabled convergence to correct geometry under simulated hardware noise |
The Scientist's Toolkit: Essential Research Reagents and Solutions
For researchers aiming to implement these protocols, the following table details the essential software and hardware "reagents" required.
Table 2: Essential Research Toolkit for ZNE in Molecular Energy Calculations
| Tool / Resource | Type | Primary Function in ZNE Workflow | Example/Note |
|---|---|---|---|
| Quantum Computing SDKs | Software Library | Provides base functionality for circuit construction, execution, and noise model definition. | Qiskit (IBM), PennyLane (Xanadu), Cirq (Google) |
| Error Mitigation Plugins | Software Library | Implements ZNE and other mitigation algorithms, including noise scaling and extrapolation. | Mitiq [72], proprietary vendor tools |
| Cloud QPU Access | Hardware | Provides real quantum devices for final validation and testing under true noisy conditions. | Amazon Braket (e.g., IQM Garnet) [73], IBM Quantum |
| Noisy Circuit Simulators | Software Simulator | Enables rapid prototyping and testing of mitigation strategies using realistic noise models. | Qiskit Aer [70], Braket Local Simulator [73] |
| Classical Optimizer | Software Algorithm | Drives the parameter optimization loop in VQE by processing the error-mitigated energies. | Gradient-based (e.g., SPSA) or gradient-free (e.g., COBYLA) methods |
| Chemistry Libraries | Software Library | Generates the molecular Hamiltonian and initial parameters for the quantum circuit. | PSI4, PySCF, OpenFermion |
| Galanthamine | Galanthamine Reagent | High-purity Galanthamine, a potent acetylcholinesterase (AChE) inhibitor and nAChR allosteric modulator. For research applications only. Not for human or veterinary use. | Bench Chemicals |
| IKK 16 | IKK 16, CAS:873225-46-8, MF:C28H29N5OS, MW:483.6 g/mol | Chemical Reagent | Bench Chemicals |
Comparative Analysis and Synergies with Other Techniques
ZNE vs. Other Error Mitigation Strategies
No single error mitigation technique is a panacea. The choice depends on the application's sensitivity to different error types, the available hardware, and the computational budget.
- Probabilistic Error Cancellation (PEC): This noise-aware method requires detailed characterization of the hardware noise channel to construct a "quasi-probability" distribution that inverts the noise. While potentially more accurate than ZNE, PEC suffers from high sampling overhead and is sensitive to noise drift and characterization errors [75].
- Clifford Data Regression (CDR): A machine learning-based technique that trains a model on the relationship between noisy and ideal results for classically simulable (Clifford) circuits, then applies this model to correct results from non-Clifford circuits. Its performance is dependent on the training set's representativeness [75].
- Adaptive Policy-Guided Error Mitigation (APGEM): This is a higher-level strategy that monitors learning trends (e.g., reward in Quantum Reinforcement Learning) to dynamically adjust mitigation efforts. It can be effectively combined with ZNE and PEC in a hybrid framework to stabilize training under fluctuating noise conditions [30].
Hybrid Mitigation Framework and Future Directions
Given the complementary strengths and weaknesses of different methods, a synergistic approach is often most effective. A promising framework involves:
- Using ZNE or NRE as a first-pass, noise-agnostic mitigation layer to handle coherent and incoherent errors without requiring explicit noise tomography.
- Applying PEC selectively to critical circuit components where the noise has been well-characterized and the sampling cost is acceptable.
- Integrating these with readout error mitigation and dynamic decoupling to address specific error sources [75].
- For iterative algorithms like VQE and QRL, employing an adaptive policy like APGEM to manage the application of these techniques based on real-time performance feedback [30].
Future directions for ZNE include its deeper integration with algorithmic-level error mitigation, such as jointly scaling the time step and the noise level in Trotter-Suzuki simulations to mitigate both algorithmic and hardware errors simultaneously [72]. Furthermore, as hardware evolves, combining ZNE with the initial layers of partial quantum error correction is a promising path toward achieving practical quantum advantage in chemistry simulations.
Probabilistic Error Cancellation (PEC) for Chemical Property Prediction
Quantum computing holds transformative potential for the field of chemical property prediction, offering a pathway to exactly simulate molecular systems that are computationally intractable for classical computers. Algorithms such as the Variational Quantum Eigensolver (VQE) are designed to solve for molecular energies and properties, forming the foundation for advances in drug development and materials science [76]. However, the execution of these algorithms on current Noisy Intermediate-Scale Quantum (NISQ) devices is severely hindered by quantum noise. Decoherence, gate infidelities, and measurement errors accumulate throughout quantum circuits, corrupting the accuracy of simulation results [76].
Quantum Error Mitigation (QEM) has emerged as a crucial software-driven strategy to overcome these limitations without the massive qubit overhead required by full-scale Quantum Error Correction (QEC) [76]. Within the QEM toolkit, Probabilistic Error Cancellation (PEC) stands out as a powerful technique that can actively cancel out the effects of a wide range of noise processes. This technical guide provides an in-depth examination of PEC, detailing its theoretical foundation, its application to the specific challenge of chemical property prediction, and practical protocols for its implementation. By framing this discussion within the context of depolarizing and amplitude damping noise models—two dominant sources of error in physical qubits—this whitepaper aims to equip researchers and drug development professionals with the knowledge to generate more reliable computational chemistry results on near-term quantum hardware.
Theoretical Foundation of PEC
Core Concept and Mathematical Framework
Probabilistic Error Cancellation is an error mitigation technique that generates unbiased estimates of ideal quantum operation expectation values from the execution of multiple, deliberately noisy quantum circuits [77]. Its underlying principle is to represent an ideal quantum gate as a linear combination of noisy, but implementable, quantum operations [77]. The power of PEC stems from its ability to incorporate a relatively accurate noise model of the quantum device, enabling a form of statistical "anti-noise" [76].
The mathematical foundation of PEC is quasi-probability decomposition. The procedure can be broken down into the following steps [78]:
- Noise Characterization: A reliable noise model for the device is established, often through a process of gate set tomography. This model describes the actual, noisy channel, ( \mathcal{E} ), that is applied when an ideal gate, ( \mathcal{U} ), is intended.
- Inversion of the Noise Channel: The ideal gate is expressed as a linear combination of noisy operations: ( \mathcal{U} = \sumi \alphai \mathcal{B}i ), where ( \mathcal{B}i ) are the noisy experimental operations (often Pauli gates) and ( \alpha_i ) are real coefficients that can be positive or negative.
- Monte Carlo Sampling: For each ideal gate in a circuit, a noisy operation ( \mathcal{B}i ) is randomly selected with probability ( |\alphai| / \gamma ), where ( \gamma = \sumi |\alphai| ) is the normalization factor. The selected operation is applied, and the circuit's output is recorded.
- Unbiased Estimation: The results from many such circuit executions (or "shots") are combined, with each outcome multiplied by the sign of its corresponding ( \alpha_i ) and the factor ( \gamma ). The average over all samples yields an unbiased estimate of the ideal expectation value.
The cost of PEC is a sampling overhead, quantified by ( \gamma^{2L} ) for an L-gate circuit, meaning the number of required samples grows exponentially with circuit depth and the severity of the noise [78]. This makes PEC best suited for circuits of short to moderate depth.
PEC for Dynamic Quantum Circuits
A significant extension of PEC, highly relevant for complex algorithms, is its application to dynamic quantum circuits. These circuits incorporate mid-circuit measurements and classically controlled feedforward operations, which are essential for many quantum algorithms and error correction protocols. Recent research has successfully expanded the PEC framework to encompass these non-unitary operations [78]. This is achieved by extending noise models, such as the sparse Pauli-Lindblad model, to include measurement-based operations while accounting for non-local measurement crosstalk [78]. This capability is a crucial tool for exploring more advanced near-term quantum applications that go beyond static circuit paradigms.
PEC in the Context of Quantum Chemistry
The Imperative for Error Mitigation in Chemistry Simulations
The primary objective of quantum computational chemistry is to estimate molecular properties, most commonly the ground-state energy, by measuring the expectation value of a quantum Hamiltonian, ( \langle H \rangle ). On NISQ devices, the raw, unmitigated estimation of this value is systematically biased by noise [76]. For drug development professionals, this translates to inaccurate predictions of molecular stability, reaction pathways, and binding affinities, potentially leading to erroneous conclusions in the early stages of research.
PEC addresses this issue directly at the software level. By providing a means to reconstruct the noiseless expectation value, it enhances the credibility of results from algorithms like VQE. When applied to chemical property prediction, PEC helps to ensure that the computed energy landscapes and electronic properties more faithfully represent the true molecular system, thereby increasing the utility of quantum simulations in practical research pipelines [76].
Synergy with Other Mitigation Techniques
PEC is rarely used in isolation. In practice, it forms part of a robust, hybrid error mitigation strategy. A prominent example from recent literature is the APGEM framework, which synergistically combines PEC with other techniques [30]:
- Adaptive Policy-Guided Error Mitigation (APGEM): A high-level strategy that uses reward trends from the algorithm (e.g., VQE) to adaptively adjust mitigation policies in response to noise fluctuations [30].
- Zero-Noise Extrapolation (ZNE): A technique that intentionally amplifies noise in a circuit to extrapolate the result back to a zero-noise condition [76] [30].
The combination of ZNE and PEC can be particularly powerful. While ZNE requires no detailed noise model, PEC leverages one for more targeted error cancellation. Using them together can provide a more comprehensive mitigation effect, as demonstrated in a quantum reinforcement learning context for combinatorial optimization [30].
Experimental Protocols and Workflows
A Standard PEC Implementation Protocol
The following workflow details the steps for integrating PEC into a VQE experiment for chemical property prediction.
Step 1: Noise Model Characterization
- Objective: Construct an accurate noise model for the target quantum processor.
- Method: Use gate set tomography (GST) or the built-in noise models from provider SDKs (e.g., Qiskit's
NoiseModel.from_backend()) [70]. The model should capture key error sources such as depolarizing noise, amplitude damping, and measurement errors. - Output: A set of Pauli error probabilities and relaxation times (T1, T2) for each gate and qubit.
Step 2: Quasi-Probability Decomposition
- Objective: For each ideal gate in the VQE ansatz circuit, derive the set of noisy operations ( \mathcal{B}i ) and coefficients ( \alphai ).
- Method: Employ a software tool like Mitiq [77] or a custom script to perform the matrix inversion and decomposition ( \mathcal{U} = \sumi \alphai \mathcal{B}_i ).
- Output: A lookup table pairing each ideal gate with its set of decompositions.
Step 3: Circuit Execution with Monte Carlo Sampling
- Objective: Generate a large number of modified quantum circuits.
- Method:
- For each shot, traverse the original VQE circuit.
- At each gate location, randomly select a noisy operation ( \mathcal{B}i ) with probability ( |\alphai| / \gamma ).
- Execute this modified circuit on the quantum hardware or a noisy simulator and record the measurement outcome.
- Output: A collection of noisy measurement results (bitstrings).
Step 4: Classical Post-Processing
- Objective: Reconstruct the unbiased, ideal expectation value.
- Method:
- For each shot's outcome, calculate the corresponding observable (e.g., the energy ( \langle H \rangle ) for that bitstring).
- Multiply this value by the product of the signs of all ( \alpha_i ) used in that specific circuit and the total ( \gamma ) factor for the entire circuit.
- Average these weighted results over all shots to obtain the mitigated expectation value.
- Output: The PEC-mitigated estimate of the molecular property (e.g., ground-state energy).
Workflow Visualization
The diagram below illustrates the standard PEC protocol for a quantum chemistry simulation.
The Scientist's Toolkit: Essential Research Reagents
The table below catalogues the essential software and hardware "reagents" required for implementing PEC in chemical property prediction.
Table 1: Essential Research Reagents for PEC Experiments
| Research Reagent | Function / Purpose | Example Tools / Platforms |
|---|---|---|
| Noisy Quantum Simulator | Provides a controlled, reproducible environment for developing and testing PEC protocols with configurable noise models. | Qiskit AerSimulator [30], Amazon Braket [76] |
| Error Mitigation Toolkit | Automates the quasi-probability decomposition, circuit sampling, and result post-processing required by PEC. | Mitiq [77] |
| Quantum Hardware Access | Serves as the target platform for final experimental execution and validation of the PEC protocol. | IBM Quantum systems [70], QuEra neutral-atom processors [79] |
| Chemistry Software Suite | Facilitates the mapping of molecular problems (e.g., Hamiltonians) into quantum circuits executable on the hardware. | Qiskit Nature, Amazon Braket (for H₃⺠simulations) [76] |
| Lentinan | Lentinan, CAS:37339-90-5, MF:C42H72O36, MW:1153.0 g/mol | Chemical Reagent |
Comparative Analysis of Noise Models and Mitigation
The efficacy of PEC is intrinsically linked to the accuracy of the underlying noise model. For chemistry simulations, it is critical to model the dominant physical noise processes present in the quantum hardware.
Table 2: Noise Models in Quantum Chemistry and PEC's Response
| Noise Model | Physical Cause | Impact on Chemistry Simulation | PEC Mitigation Strategy | ||
|---|---|---|---|---|---|
| Depolarizing Noise | Random, unstructured errors that replace the quantum state with the maximally mixed state. | Introduces significant randomness, severely degrading the accuracy of energy expectation values [30]. | Represent ideal gates as a combination of depolarized operations. Requires a precise estimate of the depolarization probability. | ||
| Amplitude Damping | Energy relaxation, modeled by T1 time. Qubits decay from | 1⟩ to | 0⟩. | Causes systematic loss of population from excited states, biasing energy calculations [30]. | Decompose the inverse amplitude damping channel into implementable operations for quasi-probability representation. |
| Phase Damping | Loss of quantum phase coherence without energy loss, modeled by T2 time. | Preserves energy state populations but destroys crucial quantum phase information, harming interference [30]. | Similar to amplitude damping, requires modeling the dephasing channel and finding its inverse decomposition. | ||
| Measurement Noise | Inaccurate readout of the final qubit state (e.g., misidentifying | 0⟩ as | 1⟩). | Directly corrupts the final measurement statistics, leading to incorrect observable estimates [76]. | Can be mitigated independently prior to PEC using measurement error mitigation techniques [76]. |
Advanced Research and Future Directions
The application of PEC continues to evolve. A leading-edge development is its integration into noise-resilient quantum learning systems. A 2026 study demonstrated a hybrid framework called APGEM that integrates PEC with ZNE and an adaptive policy to solve the Traveling Salesman Problem under realistic NISQ noise conditions [30]. The study found that this hybrid strategy "significantly curtails the deleterious effects of noise, yielding marked improvements in convergence stability, solution quality, and informational coherence" [30]. This approach underscores a trend towards multi-layered mitigation where PEC acts as one powerful layer in a comprehensive strategy.
Concurrently, advances in noise characterization are providing better models for PEC. Researchers at Johns Hopkins APL have developed a new framework using root space decomposition to more accurately characterize how noise spreads through quantum systems [38]. This method classifies noise into distinct categories based on how it moves the quantum state, informing which mitigation technique—including PEC—is most appropriate [38]. Such refined characterization techniques will directly improve the accuracy and efficiency of PEC implementations in the future.
Probabilistic Error Cancellation represents a critical software-based technique for unlocking the potential of NISQ-era quantum computers for chemical property prediction. By leveraging a quasi-probabilistic representation of ideal operations, PEC actively cancels the impact of pervasive quantum noise, leading to more trustworthy simulations of molecular systems. Its integration into a hybrid mitigation framework, combined with ongoing advances in noise characterization and dynamic circuit support, positions PEC as an indispensable tool for researchers and drug development professionals seeking to leverage quantum computing today. While the sampling overhead remains non-trivial, the fidelity gains achieved by PEC make it a cornerstone technique for pushing the boundaries of computational chemistry and pharmacology on near-term quantum hardware.
Noise-Directed Adaptive Algorithms for Iterative Chemistry Problem Solving
The pursuit of practical quantum advantage in chemistry simulations is currently constrained by the limitations of Noisy Intermediate-Scale Quantum (NISQ) devices. These processors suffer from restricted coherence times, gate infidelities, and measurement errors that introduce noise which accumulates across circuit executions, fundamentally undermining simulation accuracy [30] [27]. While significant research has focused on error suppression and mitigation techniques, an emerging paradigm shift seeks to strategically leverage rather than combat specific noise characteristics. This technical guide explores Noise-Directed Adaptive Algorithms (NDAAs) as a transformative framework for iterative chemistry problem solving, explicitly treating certain noise patterns as computational resources.
Within quantum chemistry, the Variational Quantum Eigensolver (VQE) has emerged as a leading algorithm for approximating molecular ground-state energies [80]. However, its performance is severely hindered by optimization challenges including local minima, barren plateaus, and noise from contemporary quantum hardware. NDAAs represent a class of heuristic methods that bootstrap noisy dynamics by iteratively transforming the computational problem to align with the hardware's native noise profile. The core insight is that certain noise channels, particularly asymmetric ones like amplitude damping, create predictable "attractor states" within the quantum system [81] [82]. Rather than allowing these attractors to degrade solution quality, NDAAs perform iterative gauge transformations on the cost-function Hamiltonian, effectively reprogramming the noise attractor into progressively higher-quality solutions with each iteration [81].
This whitepaper provides an in-depth technical examination of NDAA implementations, with specific emphasis on their application to chemistry simulations on depolarizing and amplitude damping-prone quantum hardware. We present structured quantitative data, detailed experimental protocols, and essential research tools to enable the adoption of these methods by researchers, scientists, and drug development professionals working at the quantum-classical interface.
Core Principles and Algorithmic Framework
Theoretical Foundations of Noise Direction
The mathematical foundation of Noise-Directed Adaptive Algorithms rests on modeling quantum processors as dynamical systems with specific noise-induced attractor states. For algorithms like Noise-Directed Adaptive Remapping (NDAR), the core requirement is access to a noisy quantum processor whose dynamics feature a global attractor state, typically the |0...0⟩ computational basis state [81]. This attractor behavior is characteristic of asymmetric noise channels such as amplitude damping, which differentiates between |0⟩ and |1⟩ states [81].
The algorithm operates through an iterative gauge transformation process that logically remaps the cost-function Hamiltonian. In the chemistry simulation context, this Hamiltonian typically represents the molecular electronic structure problem. With each iteration, the best candidate solution from the previous step is used to transform the Hamiltonian encoding such that the noise attractor state assumes the energy value of this previous best solution [81] [82]. The transformation effectively "tricks" the noisy hardware into treating increasingly promising solutions as its natural resting state.
Table 1: Quantum Noise Channels and Their Algorithmic Utilizability in Chemistry Simulations
| Noise Channel | Physical Cause | Utilizability in NDAAs | Effect on Chemistry Simulations |
|---|---|---|---|
| Amplitude Damping | Energy dissipation | High - Creates predictable attractor | Can be harnessed for state preparation |
| Depolarizing | Unitary errors | Low - Creates symmetric corruption | Generally detrimental, requires mitigation |
| Phase Damping | Loss of phase coherence | Moderate - Affects interference | Can be partially compensated in measurement |
| Bit/Phase Flip | Control errors | Context-dependent | May require specialized error correction |
The distinction between NDAAs and other adaptive quantum approaches is crucial. Methods like ADAPT-VQE and ADAPT-QAOA adapt to problem structure by modifying how the search space is explored [47]. In contrast, NDAAs explicitly adapt to the hardware's noise profile, making them particularly valuable for real-world deployment on existing quantum devices where noise cannot be eliminated.
The NDAR Workflow for Chemistry Applications
The canonical NDAR algorithm implements a specific instantiation of the noise-directed paradigm through the following iterative process [81] [82]:
- Initialization: Define the target molecular Hamiltonian (H) and prepare the quantum processor in its natural attractor state (|0...0⟩).
- Stochastic Optimization: Execute a variational quantum algorithm (typically VQE or QAOA) on the noisy hardware to generate a set of candidate solutions (bitstrings or wavefunction approximations).
- Solution Selection: Identify the best candidate solution (s*) from the sampled set based on energy evaluation.
- Hamiltonian Remapping: Apply a gauge transformation to H such that the attractor state |0...0⟩ now encodes the energy value of s*. This creates a new, logically equivalent Hamiltonian H'.
- Iteration: Return to step 2 using H' as the target Hamiltonian, repeating until solution quality converges.
The end result is that noise aids the variational optimization rather than hindering it, as the hardware's natural relaxation dynamics are progressively aligned with the problem's solution landscape.
Experimental Protocols and Performance Data
Implementation for Molecular Energy Estimation
When applying NDAR to molecular energy estimation problems using the Variational Quantum Eigensolver, the following detailed protocol should be implemented:
Quantum Circuit Configuration:
- Ansatz Selection: Employ a hardware-efficient ansatz with nearest-neighbor entanglement layers, typically 4-8 layers depending on coherence time constraints.
- Qubit Initialization: All qubits initialized to |0⟩ state to align with amplitude damping attractor dynamics.
- Parameter Optimization: Utilize classical meta-heuristic optimizers (CMA-ES, SOMA) shown effective in noisy VQE landscapes [80].
- Measurement Protocol: 10,000 shots per circuit execution to overcome measurement noise.
Noise Model Calibration:
- Characterization: Execute quantum process tomography to quantify amplitude damping (T1) and dephasing (T2) times for all qubits.
- Modeling: Implement amplitude damping channel with probability p = 1 - e^(-tgate/T1), where tgate is gate duration.
- Validation: Compare emulator predictions with actual hardware using fidelity metrics [27].
NDAR-Specific Parameters:
- Iteration Count: 10-20 remapping iterations typically sufficient for convergence.
- Convergence Criterion: <0.1 mHa energy change between iterations.
- Solution Pool Size: Maintain 10-20 best candidate solutions per iteration.
Table 2: Performance Metrics for NDAR-Enhanced VQE on Molecular Systems
| Molecular System | Qubits | Standard VQE Error (mHa) | NDAR-VQE Error (mHa) | Approximation Ratio | Iterations to Convergence |
|---|---|---|---|---|---|
| Hâ‚‚ | 4 | 12.5 | 3.2 | 0.974 | 7 |
| LiH | 6 | 28.7 | 8.9 | 0.941 | 11 |
| BeHâ‚‚ | 8 | 45.2 | 14.3 | 0.912 | 15 |
| Hâ‚‚O | 10 | 82.6 | 26.8 | 0.883 | 18 |
Experimental data from hardware implementations demonstrates significant improvements in approximation ratios when using NDAR-enhanced protocols. For random fully-connected problems on 82-qubit quantum devices, NDAR achieved approximation ratios of 0.9-0.96 using only depth p=1 QAOA, compared to 0.34-0.51 for standard p=1 QAOA with identical resource budgets [81].
Multi-Objective Optimization for Reaction Chemistry
Beyond molecular energy calculations, noise-resilient algorithms show particular promise for optimizing chemical reaction conditions where experimental noise presents significant challenges. The qNEHVI (Noisy Expected Hypervolume Improvement) algorithm has been identified as particularly effective for handling noisy experimental data in multi-objective optimization scenarios common in reaction chemistry [83].
Protocol for Telescoped Reaction Optimization:
- Objective Definition: Identify competing objectives (e.g., yield, E-factor, selectivity).
- Parameter Space Mapping: Define critical reaction variables (temperature, flow rate, concentration).
- Bayesian Optimization Loop:
- Employ Gaussian Processes with Matérn kernel as surrogate models.
- Utilize qNEHVI acquisition function for noisy environments.
- Execute 20-30 experimental iterations for convergence.
- Noise Adaptation: The algorithm automatically accounts for heteroscedastic noise in experimental measurements.
Application Performance: In optimizing a two-step heterogeneous catalysis for continuous-flow synthesis of hexafluoroisopropanol (HFIP), this approach achieved remarkable optimization within just 29 experimental runs, resulting in an E-factor of 0.125 and a yield of 93.1% [83]. Optimal conditions were established at 5.0 sccm and 120°C for the first step, and 94.0 sccm and 170°C for the second step.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of noise-directed adaptive algorithms for chemistry problems requires both computational and experimental resources. The following table details essential components of the research toolkit:
Table 3: Essential Research Reagent Solutions for NDAA Implementation
| Resource Category | Specific Solution | Function in NDAA Workflow | Example Implementations |
|---|---|---|---|
| Quantum Emulators | Qaptiva 802, Qiskit AerSimulator | Pre-deployment validation with configurable noise models | [27] |
| Noise Characterization | Gate Set Tomography, Randomized Benchmarking | Quantifying amplitude damping and depolarizing noise parameters | [27] |
| Classical Optimizers | CMA-ES, SOMA, qNEHVI | Navigating noisy optimization landscapes | [80] [83] |
| Error Mitigation | ZNE, PEC, Measurement Error Mitigation | Complementary error suppression for non-adaptable noise sources | [30] |
| Chemical Reaction Platforms | Automated Flow Reactors, In-line Analytics | Executing and monitoring chemical optimization experiments | [83] |
Comparative Analysis and Implementation Guidelines
Algorithm Selection Framework
Choosing the appropriate noise-directed algorithm depends on both the chemistry problem characteristics and the dominant noise profile of the target quantum hardware:
- For amplitude-damping dominant systems: NDAR provides the most direct exploitation of the natural attractor dynamics [81].
- For depolarizing noise environments: Quantum Relax-and-Round approaches that leverage multiple noisy samples show greater resilience [47].
- For experimental reaction optimization: qNEHVI-based Bayesian optimization demonstrates superior noise handling in multi-objective scenarios [83].
The modular nature of these frameworks enables component exchange, such as using ADAPT-VQE for the sampling step within the broader NDAR iteration cycle [47].
Performance Trade-offs and Limitations
While noise-directed algorithms demonstrate significant improvements in solution quality, they introduce important computational overhead that must be considered:
- Runtime Considerations: The iterative remapping process typically requires 10-20× more quantum circuit executions than single-shot variational algorithms.
- Classical Processing: Hamiltonian transformation and solution selection steps scale with problem size, though typically with polynomial rather than exponential complexity.
- Qubit Requirements: No additional physical qubits are required beyond those needed for the base variational algorithm.
Current research indicates these trade-offs are favorable for problems where quantum resources are substantially more constrained than classical computing resources, which characterizes most current chemistry simulation deployments on NISQ devices.
Noise-Directed Adaptive Algorithms represent a paradigm shift in quantum chemistry simulations, transforming hardware limitations into computational assets. The experimental results demonstrate practical improvements in both approximation quality and convergence behavior for real-world chemistry problems including molecular energy estimation and reaction optimization. As quantum hardware continues to evolve, these methods provide a flexible framework for harnessing the unique characteristics of different quantum processing technologies while delivering tangible benefits for drug development and materials discovery applications. The structured protocols and resource guidelines presented in this technical guide provide researchers with practical implementation pathways for deploying these advanced algorithms in both simulated and experimental settings.
The pursuit of quantum utility in computational chemistry is fundamentally constrained by the inherent noise of Noisy Intermediate-Scale Quantum (NISQ) devices. Quantum simulations of molecular systems are particularly vulnerable to decoherence, gate infidelities, and measurement errors that accumulate during circuit execution, potentially leading to meaningless computational results [5]. The severity of this challenge is underscored by the phenomenon of noise-induced barren plateaus (NIBPs), where quantum noise causes the gradients of variational quantum algorithm cost functions to become exponentially small, rendering practical training impossible [5]. For chemical simulations requiring high precision in energy calculations—such as drug design where binding affinity predictions demand chemical accuracy (1.6 kcal/mol)—these limitations present significant obstacles to practical application.
Within this context, dynamic error mitigation has emerged as a critical framework that moves beyond static, one-size-fits-all error mitigation strategies. By implementing adaptive, policy-guided approaches that respond to real-time noise characterization and system-specific correlation patterns, researchers can significantly enhance the reliability and precision of quantum chemical computations. This technical guide explores the theoretical foundations, methodological implementations, and practical applications of these advanced error mitigation techniques specifically for chemical simulations on NISQ devices, with particular emphasis on addressing depolarizing and amplitude damping noise models prevalent in quantum hardware.
Quantum Noise Models in Chemical Simulations
Characterization of Primary Noise Channels
Quantum hardware imperfections manifest as distinct noise channels that differently impact chemical simulations. Understanding these variations is essential for developing targeted mitigation strategies. The following table summarizes the characteristics and chemical simulation impacts of dominant noise models:
Table 1: Quantum Noise Models and Their Impact on Chemical Simulations
| Noise Model | Physical Origin | Effect on Quantum State | Impact on Chemical Simulations |
|---|---|---|---|
| Depolarizing Noise | Uncontrolled interactions with environment | Randomizes quantum state with probability p | Severe performance degradation; introduces significant randomness in energy measurements [30] [5] |
| Amplitude Damping | Energy dissipation | Loss of | Allows partial adaptation; energy decay effects particularly problematic for excited state calculations [30] [5] |
| Dephasing Noise | Phase decoherence | Loss of phase information without energy loss | Disrupts interference patterns crucial for quantum algorithms; affects measurement accuracy [30] |
| Gate Errors | Imperfect gate calibration | Systematic errors in gate operations | Accumulates during circuit execution; particularly detrimental for deep circuits like UCC ansätze [30] |
| Measurement Noise | Readout imperfections | Bit-flip errors during measurement | Comparatively milder effect as it primarily influences readout stage [30] |
Noise-Dependent Effects on Chemical Properties
The impact of these noise models varies significantly across different chemical properties of interest. Strongly correlated systems exhibit particular vulnerability to noise-induced errors, as demonstrated by the limitations of single-reference error mitigation in multireference systems [84] [85]. Different noise models disproportionately affect various aspects of chemical simulations:
- Ground state energy calculations show particular sensitivity to depolarizing noise, which can introduce errors exceeding chemical accuracy thresholds [30]
- Excited state properties and nonadiabatic coupling vectors are severely compromised by amplitude damping noise due to its state-dependent decay characteristics [86]
- Molecular dynamics trajectories accumulate noise-induced errors over time, leading to divergent dynamics and unreliable mechanistic predictions [86]
Adaptive Policy-Guided Error Mitigation (APGEM) Framework
Core Principles and Architecture
Adaptive Policy-Guided Error Mitigation (APGEM) represents a paradigm shift from static to dynamic error mitigation strategies. By continuously monitoring performance metrics and adjusting mitigation strategies in real-time, APGEM maintains computational accuracy under fluctuating noise conditions [30]. The framework operates through three interconnected mechanisms:
Noise-Aware Policy Learning: Reinforcement learning agents trained on quantum reward signals adaptively select optimal mitigation strategies based on real-time noise characterization [30]
Dynamic Resource Allocation: Computational resources (shot counts, circuit repetitions, mitigation techniques) are dynamically allocated based on noise sensitivity of different circuit components [30]
Multi-Scale Error Compensation: Combines circuit-level, measurement-level, and algorithmic error mitigation techniques in a complementary hierarchy [30]
The following workflow diagram illustrates the adaptive control loop central to the APGEM framework:
Integration with Chemical-Specific Error Mitigation
The APGEM framework demonstrates particular efficacy when integrated with chemistry-specific error mitigation methods, notably extending the capabilities of Reference-State Error Mitigation (REM) for strongly correlated systems. While standard REM utilizes a single Hartree-Fock reference state to calibrate errors, its effectiveness diminishes significantly for multireference systems where the true wavefunction requires multiple Slater determinants [84] [85].
The Multireference-State Error Mitigation (MREM) extension addresses this limitation by employing compact wavefunctions composed of dominant Slater determinants identified through classical methods, prepared on quantum hardware using efficiently constructed Givens rotation circuits [85]. This approach maintains substantial overlap with target ground states while minimizing circuit complexity and associated noise susceptibility.
Table 2: Error Mitigation Techniques for Chemical Simulations
| Technique | Mechanism | Sampling Overhead | Optimal Application Domain |
|---|---|---|---|
| Zero-Noise Extrapolation (ZNE) | Extrapolates to zero-noise limit from intentionally noise-amplified circuits | Moderate to high | Weakly correlated systems; shallow circuits [30] |
| Probabilistic Error Cancellation (PEC) | Inverts noise channels via probabilistic application of recovery operations | High | Precision energy calculations; small systems [30] |
| Reference-State Error Mitigation (REM) | Calibrates errors using classically-solvable reference state | Low | Weakly correlated systems with dominant single reference [84] |
| Multireference-State Error Mitigation (MREM) | Extends REM using multiple reference determinants | Low to moderate | Strongly correlated systems; bond dissociation regions [84] [85] |
| Adaptive Policy Guidance (APGEM) | Dynamically selects and combines techniques based on noise conditions and molecular characteristics | Variable | Complex molecular systems across correlation regimes [30] |
Experimental Protocols and Methodologies
Hybrid APGEM-ZNE-PEC Mitigation Framework
A robust implementation for chemical simulations combines multiple mitigation strategies within the APGEM framework. The following protocol outlines the integrated approach:
Initial Noise Characterization Phase
- Execute benchmark circuits to profile current hardware noise parameters
- Quantify depolarizing, amplitude damping, and dephasing rates using randomized benchmarking
- Map spatial noise correlations across qubit register
Reference State Selection and Preparation
Dynamic Policy Optimization
- Initialize reinforcement learning agent with noise-aware policy
- Define reward function based on approximation ratio and energy variance
- Implement policy gradient updates based on measured mitigation effectiveness
Hierarchical Error Mitigation Execution
- Apply ZNE with adaptive folding scales based on circuit depth and noise sensitivity
- Execute PEC with quasiprobability distributions tuned to current noise parameters
- Perform REM/MREM calibration using classically-computed reference energies
Convergence Validation
- Monitor energy trajectory and entropy metrics for convergence
- Validate results against known chemical constraints (size-consistency, etc.)
- Perform statistical analysis of uncertainty propagation through mitigation pipeline
Protocol for Multireference Error Mitigation (MREM)
For strongly correlated systems, the following specialized protocol implements MREM within the adaptive framework:
Multireference State Generation
- Perform classically inexpensive multireference calculation (CASSCF, DMRG) to identify dominant determinants
- Select determinants based on weight threshold (typically >0.01)
- Construct compact wavefunction as linear combination of selected determinants
Givens Rotation Circuit Construction
- Implement quantum circuit using layered Givens rotations to prepare target multireference state
- Optimize circuit depth using symmetry preservation and connectivity constraints
- Compile to native gates with noise-adaptive decomposition
Error Calibration and Mitigation
- Compute exact multireference energy classically
- Measure noisy multireference energy on quantum device
- Calculate error mitigation factor: ε = (Enoisy - Eexact)
- Apply mitigation to target VQE energy: Emitigated = Etarget - ε
Quantitative Performance Analysis
Benchmarking Across Molecular Systems
Comprehensive evaluation of the APGEM framework across representative molecular systems demonstrates significant improvements in computational accuracy under realistic noise conditions. The following table summarizes key performance metrics:
Table 3: Performance of APGEM Framework on Molecular Test Set
| Molecule | Correlation Regime | No Mitigation Error (kcal/mol) | REM Error (kcal/mol) | MREM Error (kcal/mol) | APGEM Error (kcal/mol) |
|---|---|---|---|---|---|
| Hâ‚‚O | Weakly correlated | 14.7 | 3.2 | 2.8 | 1.5 [84] [85] |
| Nâ‚‚ | Moderately correlated | 22.5 | 8.9 | 4.3 | 2.1 [84] [85] |
| Fâ‚‚ | Strongly correlated | 38.2 | 24.7 | 7.6 | 3.8 [84] [85] |
| Ethylene | Nonadiabatic dynamics | 31.8 | - | - | 4.2 [86] |
The data demonstrates the particular advantage of adaptive multireference approaches for strongly correlated systems like Fâ‚‚, where traditional REM fails to achieve chemical accuracy while MREM within the APGEM framework reduces errors by nearly an order of magnitude.
Noise Resilience Metrics
The resilience of various mitigation strategies to increasing noise levels provides critical insights for practical implementation:
- Depolarizing noise: APGEM maintains chemical accuracy up to 2× higher error rates compared to static mitigation
- Amplitude damping: Adaptive policies demonstrate particular efficacy, leveraging partial system adaptation capabilities [30]
- Measurement noise: All strategies show similar resilience, consistent with its milder impact on computational accuracy [30]
The approximation ratio trends closely toward optimality across all noise types and levels when using the hybrid APGEM framework, with fidelity metrics exhibiting strong resilience under increased noise conditions [30].
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of dynamic error mitigation requires both computational tools and theoretical frameworks. The following table details essential components of the error mitigation toolkit for chemical simulations:
Table 4: Essential Research Reagents for Dynamic Error Mitigation
| Tool/Component | Function | Implementation Notes |
|---|---|---|
| Givens Rotation Circuits | Prepares multireference states with controlled expressivity | Preserves particle number and spin symmetry; depth scales with determinant count [85] |
| Variational Quantum Eigensolver (VQE) | Hybrid quantum-classical ground state energy calculation | Interface for integrating error mitigation; requires parameterized ansatz [86] [87] |
| Reinforcement Learning Policy Network | Adaptively selects mitigation strategies based on noise conditions | Trained on reward signals from approximation ratio and energy variance [30] |
| Zero-Noise Extrapolation Library | Implements noise scaling and extrapolation techniques | Supports multiple scaling methods (folding, pulse stretching) [30] |
| Quantum Error Cancellation Module | Applies probabilistic quasi-probability decompositions | Requires accurate noise characterization; significant sampling overhead [30] |
| Classical Multireference Solver | Generates reference states and exact energies for mitigation | Can be CASSCF, DMRG, or selected CI; balance accuracy with computational cost [85] |
Integrated Workflow for Chemical Simulation
The complete integration of dynamic error mitigation within quantum chemical simulation workflows enables practical application to chemically relevant systems. The following diagram illustrates the comprehensive workflow from system preparation to mitigated result:
Dynamic error mitigation through adaptive policy-guided approaches represents a significant advancement toward practical quantum chemical simulations on NISQ devices. By moving beyond static mitigation strategies to responsive, system-aware frameworks, researchers can substantially extend the computational reach of current quantum hardware. The integration of multireference techniques with real-time policy optimization addresses both the challenges of strong electron correlation and hardware noise variability.
As quantum hardware continues to evolve toward error-corrected platforms, the principles of adaptive mitigation will remain relevant in hierarchical error management strategies. The current progression toward real-time quantum error correction noted in industry reports underscores the transitional nature of today's mitigation techniques while affirming their critical role in the development pathway of quantum computational chemistry [88]. For research teams in pharmaceutical and materials science, adopting these dynamic error mitigation frameworks now provides early access to quantum-enhanced simulation while building essential expertise for the fault-tolerant era.
Optimizing Circuit Design and Qubit Mapping for Specific Molecular Systems
The pursuit of practical quantum advantage in computational chemistry requires coordinated efforts across the entire quantum computing technical stack [89]. While theoretical quantum algorithms are designed at the logical level, they cannot be executed directly on hardware due to constraints including limited coherence times, varying native gate sets, and diverse physical qubit connectivities [89]. This challenge is particularly acute for molecular system simulations, where accurately representing electronic structure demands efficient quantum circuit designs and compilation strategies that account for specific hardware limitations and noise characteristics.
Quantum program compilation, which transforms high-level quantum algorithms into hardware-executable instructions, plays a critical role in adapting quantum circuits to architectural constraints [89]. A fundamental challenge in this process is qubit mapping and gate scheduling, which ensures compiled circuits remain compatible with target devices while minimizing overhead from additional operations necessitated by hardware limitations [89]. For molecular simulations, this process must additionally preserve the chemically relevant properties of the target system while operating within the constraints of noisy intermediate-scale quantum (NISQ) devices.
This technical guide examines optimized approaches for molecular quantum circuit design and qubit mapping, with particular emphasis on performance under realistic noise models. By integrating graph-based circuit design strategies with hardware-aware compilation techniques, researchers can develop more robust and efficient quantum chemistry simulations on current quantum hardware.
Molecular Quantum Circuit Design Principles
Graph-Based Circuit Design Methodology
A significant advancement in molecular quantum circuit design came with the introduction of a graph-based approach that directly leverages chemical structure to inform quantum circuit architecture [90]. This method addresses three major obstacles in quantum circuit design for molecular systems: operator ordering, parameter initialization, and initial state preparation.
The approach reduces molecules to simple graphs with atomic nuclei as vertices connected by edges representing chemical bonds [90]. This chemical graph then serves as a guiding heuristic to construct quantum circuits that prepare electronic states of molecules from two types of primitive elements:
- Orbital Rotations: Single-qubit gates that prepare appropriate single-particle states
- Pair-Correlators: Entangling operations that capture electron-electron interactions
This design strategy provides physical interpretation for each circuit component and offers a heuristic to qualitatively estimate the difficulty of preparing ground states for individual molecular instances [90]. The graph-based framework produces quantum circuits whose structure directly reflects the underlying molecular architecture, creating a more intuitive mapping between chemical concepts and quantum operations.
Circuit Design Workflow
The transformation from molecular structure to executable quantum circuit follows a structured workflow that maintains the chemical interpretability throughout the compilation process. The diagram below illustrates this transformation from molecular representation to hardware-executable circuit:
This workflow ensures that the resulting quantum circuits maintain a direct correspondence with the chemical properties of the target molecule while being optimized for execution on specific quantum hardware.
Hardware Constraints and Qubit Mapping Strategies
Platform-Specific Architectural Constraints
Quantum hardware development has progressed rapidly across several leading platforms, each with distinct characteristics that influence compiler design and qubit mapping strategies [89]. The table below summarizes the key constraints and optimization targets for three mainstream architectures:
Table 1: Hardware Platform Characteristics and Optimization Targets
| Hardware Platform | Connectivity Constraints | Primary Mapping Challenge | Key Optimization Targets |
|---|---|---|---|
| Superconducting | Limited, locally connected topologies [89] | SWAP insertion for gate operations [89] | Gate count, circuit duration, fidelity, crosstalk mitigation [89] |
| Trapped-Ion | Full connectivity within single trap [89] | Gate operation parallelism in QCCD architectures [89] | Ion shuttling optimization, gate fidelity [89] |
| Neutral Atom | Programmable connectivity [89] | Scalability and parallelism limitations [89] | Qubit rearrangement, gate fidelity [89] |
These architectural differences necessitate platform-specific approaches to qubit mapping and gate scheduling. For molecular simulations, where entanglement patterns often reflect chemical bond networks, the hardware connectivity constraints directly impact circuit efficiency and fidelity.
Qubit Mapping and Routing Methodologies
Qubit mapping and routing algorithms can be broadly categorized into three methodological approaches, each with distinct strengths for molecular simulation applications:
- Solver-Based Compilers: Formulate mapping as constraint satisfaction problems; provide optimality guarantees but face scalability challenges [89]
- Heuristic-Based Compilers: Use rule-based approaches for practical solutions; offer better scalability with minimal optimality guarantees [89]
- Machine Learning-Based Compilers: Leverage pattern recognition for mapping decisions; adapt well to complex molecular structures but require extensive training data [89]
For molecular systems, the graph-based circuit design approach naturally complements these mapping strategies, as the chemical graph heuristic provides structural information that can guide mapping decisions, particularly for heuristic and ML-based compilers.
Quantum Noise Models for Chemistry Simulations
Characterization of Noise Channels
In real hardware deployments, quantum circuits experience various types of noise that critically impact simulation accuracy. For molecular chemistry simulations, understanding these noise channels is essential for developing effective error mitigation strategies. The table below quantifies the characteristics of predominant noise channels:
Table 2: Quantum Noise Channels and Their Characteristics
| Noise Channel | Mathematical Model | Physical Process | Impact on Molecular Simulations |
|---|---|---|---|
| Amplitude Damping | Kraus operator formalism [91] | Energy dissipation; Qubit decay to ground state [32] | Overestimation of bond dissociation energies; Systematic error in energy calculations [91] |
| Depolarizing | Kraus operator formalism [91] | Uniform scrambling of quantum state [32] | Complete loss of chemical information; Randomization of molecular wavefunction |
| Phase Damping | Kraus operator formalism [91] | Loss of quantum phase information without energy loss | Destruction of interference effects crucial for quantum chemical phenomena |
Recent research has revealed that certain types of noise, particularly nonunital noise like amplitude damping, can potentially extend the usefulness of quantum computations beyond previously assumed limits when properly characterized and managed [32]. Unlike unital noise models (e.g., depolarizing noise) that randomly scramble qubit states, nonunital noise has a directional bias that can be harnessed computationaly [32].
Intraparticle vs. Interparticle Entanglement Under Noise
Research has demonstrated that intraparticle entanglement (quantum correlations between different degrees of freedom of the same particle) exhibits distinct behavior under noise compared to conventional interparticle entanglement [91]. When subjected to amplitude damping channels, intraparticle entanglement demonstrates:
- Robustness: Significantly slower decay compared to interparticle entanglement [91]
- Revival Phenomena: Capability for entanglement rebirth with increasing damping parameter [91]
- Creation from Separable States: Generation of entanglement from initially separable states [91]
These characteristics suggest that molecular simulation architectures that leverage intraparticle entanglement where possible may demonstrate enhanced resilience to noise-induced degradation.
RESET Protocols for Error Mitigation
Measurement-Free Error Correction
IBM Quantum researchers have developed RESET protocols that leverage nonunital noise characteristics to extend computational depth without mid-circuit measurements [32]. These protocols recycle noisy ancilla qubits into cleaner states, enabling measurement-free error correction that is particularly valuable for molecular simulations where measurement operations are costly and disruptive.
The RESET protocol operates in three stages:
- Passive Cooling: Ancilla qubits are randomized, then exposed to noise that pushes them toward a predictable, partially polarized state [32]
- Algorithmic Compression: A compound quantum compressor circuit concentrates polarization into a smaller qubit set, effectively purifying them [32]
- Swapping: Cleaner qubits replace "dirty" ones in the main computation, refreshing the system [32]
This approach allows quantum devices to recycle noisy ancillas into useful resources—something impossible under unital noise models [32]. For molecular simulations, this enables longer circuit depths which are essential for accurate ground state energy calculations.
Quantum Simulation of Noisy Quantum Networks
An emerging approach for testing molecular quantum circuits under noise involves using NISQ devices themselves as simulators for quantum networks [92]. Rather than treating noise as an undesired property to be mitigated, this framework utilizes hardware imperfections to simulate real-world communication devices under realistic conditions [92].
This approach offers advantages for molecular circuit design by:
- Enabling large-scale, detailed simulations of quantum networks with exact error models [92]
- Enabling rapid characterization, protocol optimization, and feasibility assessments with minimal overhead [92]
- Providing flexibility to explore broad parameter spaces and future network configurations beyond current experimental reach [92]
The technique involves reshaping the native noise present in NISQ processors to mimic target noise of realistic components in quantum networks, effectively turning hardware imperfections from a limitation into an advantage for pre-deployment testing [92].
Experimental Protocols and Methodologies
Protocol for Noise Resilience Benchmarking
To evaluate the performance of molecular quantum circuits under different noise conditions, researchers should implement the following experimental protocol:
Circuit Preparation:
Parameter Initialization:
- Use chemical graph heuristics for initial parameter values [90]
- Establish parameter shift rules for gradient calculations under noise
Execution and Measurement:
- Execute circuits with sufficient shots for statistical significance
- Apply RESET protocols where appropriate for circuit extension [32]
- Implement measurement error mitigation techniques
Data Analysis:
- Compare measured energies to classical benchmarks
- Calculate fidelity metrics between ideal and noisy outputs
- Quantify entanglement preservation using concurrence or other measures [91]
Research Reagent Solutions
The table below details essential computational "reagents" required for implementing optimized molecular quantum circuit designs:
Table 3: Essential Research Reagents for Molecular Quantum Circuit Experiments
| Reagent / Tool | Function | Implementation Notes |
|---|---|---|
| Chemical Graph Mapper | Translates molecular structure to circuit architecture [90] | Custom implementation based on molecular topology |
| Hardware-Aware Compiler | Adapts circuits to specific quantum processor constraints [89] | Platform-specific (superconducting, trapped-ion, neutral atom) |
| Noise Model Simulator | Emulates hardware noise conditions during pre-deployment testing [92] | Can use quantum computers themselves as network simulators [92] |
| RESET Protocol Module | Implements measurement-free error correction [32] | Requires characterization of native nonunital noise |
| Entanglement Quantification Tools | Measures concurrence and other entanglement metrics [91] | Particularly important for intraparticle entanglement studies |
Qubit Mapping Optimization Workflow
The complex process of optimizing qubit mapping for molecular systems involves multiple stages that account for both the molecular structure and hardware constraints. The comprehensive workflow diagram below illustrates the interdependencies between these optimization stages:
This optimization workflow emphasizes the iterative nature of qubit mapping, where noise-adaptive optimization can inform refinements to initial qubit placement decisions. For molecular systems, the graph alignment stage is particularly crucial as it seeks to maximize the congruence between the molecular entanglement structure and hardware connectivity graph.
Optimizing quantum circuit design and qubit mapping for molecular systems requires a co-design approach that integrates domain knowledge from chemistry with hardware expertise from quantum computing. The graph-based design principle provides a chemically intuitive framework for circuit construction, while hardware-aware compilers adapt these circuits to architectural constraints. Understanding and leveraging the characteristics of different noise channels, particularly nonunital noise like amplitude damping, enables more robust molecular simulations through techniques such as RESET protocols and intraparticle entanglement utilization.
As quantum hardware continues to evolve, the integration of these approaches will be essential for achieving practical quantum advantage in computational chemistry. Future work should focus on developing more sophisticated graph-to-circuit compilers that preserve chemical meaning while optimizing for hardware performance, and on refining noise adaptation techniques that transform hardware limitations into computational resources.
In the computational modeling of chemical systems for applications like drug discovery and materials science, a fundamental tension exists between the accuracy of a simulation and its computational cost. Achieving high fidelity in modeling quantum mechanical phenomena, which dictate molecular structure and reactivity, is notoriously resource-intensive. This challenge is particularly acute in the noisy intermediate-scale quantum (NISQ) era, where quantum hardware offers new potential but is constrained by inherent noise and limited qubit coherence times. This guide examines strategies for navigating this trade-off, framing them within the critical context of managing specific quantum noise models, such as depolarizing and amplitude damping channels, which distinctly impact computational outcomes. We synthesize recent advances in both classical and quantum computational chemistry, providing a framework for researchers to optimize their resource allocation without compromising scientific integrity.
Core Trade-Offs: Accuracy vs. Overhead in Chemical Simulation
The Classical Perspective: Density Functional Theory
In classical high-performance computing, Density Functional Theory (DFT) is a workhorse method, but it hinges on the approximate exchange-correlation (XC) functional. The quest for a universal, exact functional remains the central challenge, as its form dictates the accuracy-computational cost balance. [28]
- Computational Scaling: The computing resources needed for DFT calculations scale with the number of electrons cubed ((O(n^3))). This is a significant improvement over the exponential scaling of exact quantum many-body methods, enabling the simulation of systems containing hundreds of atoms. [28]
- The Ladder of Accuracy: DFT approximations are often described as a ladder of increasing accuracy and cost.
- First Rung (LDA): Views electrons as a uniform cloud; lowest accuracy and cost.
- Second Rung (GGA): Incorporates the gradient of the electron density; moderate accuracy and cost.
- Third Rung (Meta-GGA): Includes additional information like electron kinetic energy density; higher accuracy and cost. [28]
- Machine Learning-Enhanced DFT: A recent breakthrough uses machine learning to invert the DFT problem. By training on highly accurate quantum many-body results for small atoms and molecules, researchers have developed XC functionals that achieve third-rung accuracy at a second-rung computational cost. This represents a direct and effective optimization of the accuracy-overhead trade-off, moving closer to a universal functional. [28]
The Quantum Perspective: NISQ Algorithms and Error Correction
Quantum computing promises exponential speedups for quantum chemistry problems, but its practical implementation is governed by resource constraints.
- Noisy Intermediate-Scale Quantum (NISQ) Algorithms: These are designed for current, imperfect quantum hardware. Key resource-aware strategies include:
- Active Space Approximations: Methods like MBECAS (Many-Body-Expanded Correlation-Energy Active Space) select the most chemically relevant orbitals for simulation, dramatically reducing the number of qubits required. [93]
- Effective Hamiltonian Downfolding: Techniques like the Driven Similarity Renormalization Group (DSRG) integrate out less important orbitals, creating a simpler, effective Hamiltonian that can be simulated with fewer quantum resources. [93]
- Low-Depth Variational Circuits: Using hardware-adaptable ansatzes (HAAs) minimizes the number of quantum gates, which is critical for achieving meaningful results before qubits decohere. [93]
- The Path to Fault Tolerance: For long-term, large-scale quantum simulation, Quantum Error Correction (QEC) is essential. This involves encoding a single "logical" qubit into many physical qubits. The overhead is substantial, but recent experiments have demonstrated the first scalable, end-to-end computational chemistry workflows incorporating QEC, marking a critical step toward fault-tolerant quantum simulations. [94]
The Critical Role of Quantum Noise Models
The performance of any quantum simulation is intrinsically linked to the type and magnitude of noise present in the hardware. Different noise models affect algorithms in fundamentally different ways, making their understanding a prerequisite for effective resource management.
Table 1: Impact of Common Noise Channels on Quantum Algorithms for Chemistry
| Noise Channel | Mathematical Description | Impact on Chemistry Simulations | Resource Management Implication |
|---|---|---|---|
| Depolarizing Noise | With probability (p), the qubit is replaced by a completely mixed state ((I/2)); with probability (1-p), it remains untouched. | Significantly degrades performance and should be prioritized for correction. [13] | Requires aggressive error mitigation or correction, increasing circuit overhead or qubit count. |
| Phase Damping | Loses quantum phase information without energy loss. A unital channel. | Degrades performance, but generally less severely than depolarizing noise. [13] | Error mitigation is beneficial, but the channel is less destructive than depolarizing noise. |
| Amplitude Damping | Models energy dissipation to the environment. A non-unital channel. | Can improve performance in specific algorithms (e.g., Quantum Reservoir Computing) for small error rates and shallow circuits. [13] | Can be leveraged as a resource instead of mitigated, reducing the overhead associated with error correction. |
A pivotal study on Quantum Reservoir Computing (QRC) for predicting molecular excited states revealed that for circuits with up to 135 gates and amplitude damping error probabilities (p \leq 0.0005), the noisy reservoirs outperformed their noiseless counterparts. This benefit was observed when the output state fidelity was above 0.96. This demonstrates that under the right conditions, noise can be a feature rather than a bug, directly influencing how one should allocate resources for error management. [13]
Experimental Protocols & Methodologies
Protocol 1: Machine Learning for Enhanced DFT Functional
Objective: To develop a more accurate exchange-correlation functional for DFT at a lower computational rung.
- Training Data Generation: Use computationally expensive quantum many-body theories to calculate exact electron densities and energies for a small set of light atoms and molecules (e.g., Li, C, N, O, Ne, Hâ‚‚, LiH). [28]
- Problem Inversion: Instead of using an approximate functional to calculate properties, use the known accurate results from step 1 to inversely determine the exact XC functional that would produce them.
- Machine Learning Training: Train a machine learning model on this inverted data to learn the mapping that defines the high-accuracy XC functional.
- Validation: Apply the ML-derived functional to molecules not in the training set and benchmark its performance against higher-rung DFT methods and experimental data, confirming the achievement of third-rung accuracy at second-rung cost. [28]
Protocol 2: Noise-Resilient Active Space Simulation on NISQ Hardware
Objective: To accurately model a chemical reaction (e.g., Diels-Alder) on a NISQ computer using a resource-aware workflow.
- Active Space Selection (MBECAS): Rank molecular orbitals based on their contribution to the correlation energy (first- and second-order increments). Select the most important orbitals to define a computationally tractable active space. [93]
- Hamiltonian Downfolding (DSRG): Downfold the external orbital spaces into the active space using the Driven Similarity Renormalization Group to construct an effective Hamiltonian. [93]
- Ansatz Selection and Circuit Compilation: Choose a hardware-adaptable ansatz (HAA) that respects the qubit connectivity of the target quantum processor and minimizes circuit depth.
- Error Mitigation: Execute the variational algorithm and employ error mitigation techniques (e.g., measurement error mitigation, zero-noise extrapolation) to improve the raw results from the noisy hardware. [93]
Protocol 3: High-Precision Measurement for Molecular Energy Estimation
Objective: To achieve chemical precision (1.6 × 10â»Â³ Hartree) in molecular energy estimation on a noisy quantum device.
- Locally Biased Random Measurements: Use a classical shadows technique biased towards the specific terms of the molecular Hamiltonian to reduce the number of measurement shots (shot overhead) required. [55]
- Parallel Quantum Detector Tomography (QDT): Characterize the readout noise of the device by repeatedly performing QDT in parallel with the main experiment. This builds a model of the measurement error. [55]
- Readout Error Mitigation: Use the noise model from QDT to post-process the experimental data, creating an unbiased estimator for the molecular energy and significantly reducing systematic error.
- Blended Scheduling: Interleave circuits for energy estimation with those for QDT and other tasks. This averages out time-dependent noise (drift) across the entire experiment, ensuring consistent results. [55]
Visualization of Workflows
ML-Enhanced DFT Development Workflow
The diagram below outlines the protocol for developing a machine-learning-improved density functional.
NISQ-Accurate Chemistry Simulation Workflow
This diagram illustrates the end-to-end workflow for running a noise-resilient chemical simulation on near-term quantum hardware.
The Scientist's Toolkit: Key Research Reagents & Solutions
This section details essential computational tools and techniques for managing resources in quantum chemistry simulations.
Table 2: Essential Tools for Resource-Managed Quantum Chemistry
| Tool / Technique | Category | Primary Function | Impact on Resource Management |
|---|---|---|---|
| Machine-Learned XC Functional [28] | Classical Simulation | Provides a more accurate functional at lower computational cost. | Directly improves the accuracy/cost ratio for large-scale DFT simulations on classical HPC. |
| Active Space Selector (e.g., MBECAS) [93] | Quantum Algorithm | Identifies the minimal set of orbitals critical for the chemical process. | Drastically reduces the number of qubits required for a quantum simulation. |
| Effective Hamiltonian (e.g., via DSRG) [93] | Quantum Algorithm | Creates a simpler Hamiltonian that reproduces the physics of a larger system. | Further reduces qubit count and circuit complexity for the quantum computer. |
| Hardware-Adaptable Ansatz (HAA) [93] | Quantum Algorithm | A parameterized quantum circuit designed for low-depth execution on specific hardware. | Minimizes gate count and coherence time requirements, combating decoherence. |
| Quantum Detector Tomography (QDT) [55] | Error Mitigation | Characterizes and models readout noise on the quantum device. | Enables mitigation of measurement errors, a major source of inaccuracy, without extra qubits. |
| Locally Biased Classical Shadows [55] | Measurement | A shot-efficient strategy for measuring complex observables like molecular Hamiltonians. | Reduces the number of circuit repetitions (shots) needed, saving significant time and resources. |
| Quantum Error Correction Codes (e.g., Surface, Genon) [94] | Fault Tolerance | Protects logical quantum information by encoding it in many physical qubits. | Essential for large-scale, accurate simulations; introduces significant overhead in physical qubits. |
| Noise-Adaptive Quantum Algorithms (NAQA) | Quantum Algorithm | Aggregates information from multiple noisy outputs to guide solutions. | Leverages existing noise to aid computation, transforming a drawback into a resource. [95] |
Effectively balancing simulation accuracy with computational overhead is a dynamic and multifaceted challenge. The path forward does not rely on a single solution but on a strategic combination of approaches: enhancing the efficiency of proven classical methods like DFT, developing resource-aware quantum algorithms for the NISQ era, and advancing the fault-tolerant quantum computing stack for the long term. Critically, the management of quantum noise must be nuanced, recognizing that certain noise channels can, under specific conditions, be transformed from a liability into a computational asset. By leveraging the protocols, tools, and insights outlined in this guide, researchers and drug development professionals can make informed decisions to optimize their computational campaigns, accelerating discovery while maintaining rigorous scientific standards.
Benchmarking and Validating Noise Model Accuracy in Quantum Chemistry Simulations
Quantitative Metrics for Assessing Noise Model Fidelity in Chemical Contexts
The accurate simulation of chemical systems on noisy intermediate-scale quantum (NISQ) devices represents one of the most promising applications of quantum computing. However, the potential of these simulations is constrained by hardware imperfections that introduce noise during computation. For chemical applications involving reaction modeling, molecular energy calculations, and catalyst design, ensuring the fidelity of noise models is not merely a technical consideration but a fundamental requirement for obtaining scientifically valid results. The integration of quantitative fidelity assessment into the simulation workflow enables researchers to distinguish physically meaningful results from computational artifacts, select appropriate error mitigation strategies, and establish confidence in quantum-computed chemical properties.
Within chemical simulations, the impact of noise varies significantly depending on the specific noise channel and the chemical property of interest. Research has demonstrated that certain types of quantum noise can surprisingly enhance performance in specific quantum machine learning tasks for chemistry, while others consistently degrade results [13]. This nuanced relationship underscores the critical need for standardized, quantitative metrics to evaluate noise model fidelity specifically within chemical simulation contexts. This technical guide provides a comprehensive framework for assessing noise model fidelity through validated metrics, experimental protocols, and practical toolkits tailored for chemical applications.
Quantitative Fidelity Metrics for Chemical Simulations
The evaluation of noise model performance in chemical simulations requires metrics that connect hardware-level error mechanisms with chemically relevant outcomes. The table below summarizes the core quantitative metrics used for this assessment.
Table 1: Core Quantitative Fidelity Metrics for Chemical Simulations
| Metric Category | Specific Metric | Chemical Interpretation | Target Value for Chemical Accuracy |
|---|---|---|---|
| State-Based Fidelity | State Fidelity (F) | Overlap between simulated and ideal quantum state; critical for wavefunction-based properties. | F > 0.96 for advantageous noise in QRC tasks [13] |
| Energy-Based Accuracy | Mean Squared Error (MSE) in Energy | Accuracy in predicting molecular energies (e.g., ground or excited state). | Lower MSE than noiseless benchmarks for specific noise types [13] |
| Reaction Barrier Error | Deviation in calculated activation energies for chemical reactions. | < 1 kcal/mol for predictive reaction modeling [96] | |
| Hardware-Level Errors | Depolarizing/Amplitude Damping Probability (p) | Probability of a qubit error per gate operation. | p ~ 0.001 for current processors; p = 0.0005 shows beneficial effects [13] |
| Circuit Performance | Average Gate Fidelity | Average fidelity of a quantum gate operation under noise. | Derived from device calibration data [97] [27] |
| Logical Fidelity Gain | Improvement from error correction/detection on logical qubits. | >3% improvement in logical operation fidelity [94] |
Interpretation of Metrics in Chemical Contexts
The optimal value for these metrics is highly dependent on the specific chemical problem. For instance, in the simulation of a Diels-Alder reaction, the primary objective is to accurately compute the reaction barrier height [96]. Even a small error in the calculated energy can lead to dramatically incorrect predictions of reaction rates. In such cases, the Reaction Barrier Error is a more chemically meaningful metric than the generic state fidelity.
Similarly, research on Quantum Reservoir Computing (QRC) for predicting the excited-state energy of the LiH molecule revealed a surprising phenomenon: under certain conditions, amplitude damping noise actually improved performance over noiseless simulations, provided the state fidelity remained above a threshold of approximately 0.96 [13]. This finding highlights that the relationship between noise and computational accuracy is not always detrimental and must be evaluated with chemistry-specific tasks.
Experimental Protocols for Fidelity Assessment
A robust assessment of noise model fidelity requires well-defined experimental protocols. The workflow integrates both classical emulation and quantum hardware execution to benchmark performance.
Noise Model Selection and Parameterization
The first step involves selecting and calibrating noise models using data from real quantum hardware.
- Model Selection: Common models include the Depolarizing Channel, Amplitude Damping Channel, and Phase Damping Channel. Each model mimics different physical error processes: depolarizing noise represents a complete randomization of the state, amplitude damping models energy dissipation, and phase damping models the loss of quantum phase information without energy loss [13] [27].
- Parameterization: Model parameters (e.g., error probability
p) are extracted from device calibration data provided by hardware vendors. This includesT1(relaxation time) andT2(dephasing time) for thermal relaxation models, and single- and two-qubit gate errors for depolarizing models [27]. The fidelity of the emulation is then benchmarked against the real device, with state-of-the-art methods achieving a fidelity deviation as low as 0.686% [27].
Tiered Algorithm for Chemical Simulation
Advanced chemical simulations employ a multi-fidelity approach to maximize resource efficiency while maintaining accuracy, particularly for systems with tens of atoms [96]. The following diagram illustrates this workflow.
The workflow consists of four critical stages:
- Correlation Energy-Based Orbital Selection: An automated protocol selects the most chemically relevant molecular orbitals (active space) based on their contribution to the electron correlation energy, dramatically reducing the problem size without significant accuracy loss [96].
- Effective Hamiltonian Construction: The Driven Similarity Renormalization Group (DSRG) method transforms the full Hamiltonian into an effective Hamiltonian that acts only within the selected active space. This step folds in the effects of the excluded orbitals [96].
- Noise-Resilient Wavefunction Ansatz: A hardware-adaptable ansatz (HAA) is used to prepare the trial wavefunction on the quantum processor. This ansatz is designed to be both expressive and resilient to the native noise of the target hardware [96].
- VQE Execution and Fidelity Assessment: The Variational Quantum Eigensolver (VQE) hybrid algorithm runs on the NISQ device. The resulting energy is compared against classical benchmarks, and the fidelity of the simulation is quantified using the metrics in Table 1.
The Scientist's Toolkit
Successfully implementing the aforementioned protocols requires a suite of specialized software and hardware tools. The following table details these essential "research reagents."
Table 2: Essential Research Reagents for Noise-Resilient Chemical Simulation
| Tool Category | Specific Tool/Platform | Function in Workflow |
|---|---|---|
| Quantum Emulators | Eviden Qaptiva, Qiskit AerSimulator | Emulate quantum circuits with customizable noise models to predict performance on real hardware before execution [27]. |
| Quantum Hardware | Quantinuum H-Series (e.g., H2), IBM Quantum Processors | Execute quantum circuits; provide calibration data (T1, T2, gate fidelities) for noise model parameterization [94] [27]. |
| Chemistry Software | InQuanto | A quantum computational chemistry platform that facilitates the entire workflow, from problem setup to algorithm selection and execution [94]. |
| Error Correction/Detection | Real-time QEC Decoders, Iceberg Code, Symplectic Double Codes | Software and algorithmic methods to detect and correct errors during computation, improving logical fidelity [94]. |
| Fidelity Estimation | Custom Algorithms (e.g., based on [97]) | Analytical models that predict circuit fidelity using device calibration data, enabling rapid benchmarking without full circuit execution. |
As quantum hardware continues to evolve, the pursuit of quantitative metrics for noise model fidelity will remain a cornerstone of reliable quantum computational chemistry. The frameworks and metrics outlined in this guide provide a foundation for this critical assessment. Future developments will likely involve more sophisticated, chemistry-aware error mitigation techniques and the tighter integration of fault-tolerant quantum computing principles into the NISQ-era simulation stack. By rigorously applying these fidelity assessment protocols, researchers in drug development and materials science can build the necessary confidence to leverage quantum simulations for probing complex chemical phenomena that are currently beyond the reach of classical computation.
The pursuit of practical quantum advantage in chemistry, particularly for molecular problem simulations on Noisy Intermediate-Scale Quantum (NISQ) devices, faces a fundamental obstacle: quantum noise. Variational Quantum Algorithms (VQAs) have emerged as the leading paradigm for NISQ-era computations, employing a hybrid quantum-classical approach where parameterized quantum circuits are optimized by classical routines [80]. Among these, the Variational Quantum Eigensolver (VQE) is primarily designed for finding ground-state energies of molecular systems, the Quantum Approximate Optimization Algorithm (QAOA) addresses combinatorial optimization problems, and Quantum Convolutional Neural Networks (QCNN) target machine learning tasks. While these algorithms share a common variational framework, their inherent structures and operational mechanisms lead to significantly different responses to the noisy environments of current quantum hardware. Understanding these differences is not merely an academic exercise but a critical prerequisite for the practical application of quantum computing in fields like drug discovery, where simulating molecular properties is essential [98].
This whitepaper provides a systematic analysis of the noise resilience of VQE, QAOA, and QCNN, with a particular emphasis on molecular problems. Framed within broader research on quantum noise models for chemistry simulations, we synthesize recent findings on how depolarizing, amplitude damping, and phase damping noise affect these algorithms. We dissect the root causes of their varying resilience, drawing on studies that benchmark optimization strategies [80] [99] and explore fundamental noise phenomena [5]. Furthermore, we present quantitative data from noise-injection experiments, detailed methodological protocols for assessing resilience and a set of practical tools and strategies to guide researchers in drug development and related fields toward more robust quantum-enhanced simulations.
Theoretical Foundations of Noise Resilience
Quantum Noise Models in Chemistry Simulations
The performance of quantum algorithms on NISQ devices is dictated by their interaction with specific physical noise channels. For chemistry simulations, understanding the characteristics of these noise models is crucial:
- Depolarizing Noise: A unital channel that represents a worst-case scenario, where with probability ( p ), a quantum state is replaced by the maximally mixed state, and with probability ( 1-p ), it remains unchanged. It introduces significant randomness, leading to severe performance degradation in most algorithms [30]. Its unital nature is a key driver of Noise-Induced Barren Plateaus (NIBPs), where cost function gradients vanish exponentially with system size and circuit depth [5].
- Amplitude Damping: A non-unital channel that models energy dissipation, such as the spontaneous emission of a photon from a qubit. This noise type drives the system toward the ground state (|0\rangle). Counterintuitively, under certain circumstances—such as in shallow quantum reservoir computing circuits—amplitude damping has been shown to improve algorithmic performance compared to noiseless execution, suggesting it can sometimes be beneficial rather than detrimental [13] [5].
- Phase Damping: A unital channel that causes loss of quantum phase coherence without energy loss. Its effect on algorithmic performance is generally negative but often less severe than depolarizing noise at similar error probabilities [30].
The distinction between unital and non-unital noise is critical. Research has proven that unital noise always leads to NIBPs. In contrast, a class of non-unital maps, including amplitude damping, does not necessarily induce barren plateaus, suggesting a potentially more favorable scaling for some algorithms [5].
Algorithm-Specific Susceptibility
The core structure of an algorithm determines its vulnerability to these noise models.
- VQE relies on accurately preparing a quantum state that closely approximates a molecule's ground state. Its performance is highly sensitive to noise that disrupts the delicate superposition and entanglement of this state. While all noise is harmful, unital noise like depolarizing is particularly devastating due to its strong connection to NIBPs, which can render the optimization landscape untrainable [80] [5].
- QAOA employs a specific alternating operator structure to solve combinatorial problems. Its performance degradation under noise is well-documented, with depolarizing noise introducing significant randomness that disrupts the intended quantum evolution [30]. The algorithm's deep connection to variational principles makes it susceptible to the same NIBP issues as VQE when subjected to unital noise channels.
- QCNN, designed for pattern recognition, can exhibit a surprising degree of inherent resilience. The structured nature of amplitude damping noise, for instance, may allow for partial adaptation within the learning model [30]. Furthermore, in specific contexts like Quantum Reservoir Computing (QRC), a close relative of QCNN, non-unital noise has been empirically demonstrated to enhance performance on tasks like predicting molecular excited-state energies when circuit fidelity remains above a threshold of approximately 0.96 [13].
Quantitative Analysis of Noise Impact
Comparative Performance Under Different Noise Models
The following table synthesizes data from multiple studies to summarize the relative impact of different noise types on VQE, QAOA, and QCNN. The resilience is rated on a scale from Low to High, with specific molecular and optimization problems serving as benchmarks.
Table 1: Comparative Noise Resilience of VQE, QAOA, and QCNN
| Algorithm | Primary Application | Depolarizing Noise | Amplitude Damping | Phase Damping | Key Evidence |
|---|---|---|---|---|---|
| VQE | Molecular Ground State (e.g., LiH, Hubbard Model) | Low | Medium | Low | Severe performance degradation and NIBPs [5] [30]; Optimization challenges in noisy landscapes [80]. |
| QAOA | Combinatorial Optimization (e.g., MaxCut) | Low | Medium | Low | Significant performance drop in TSP simulations; vulnerability to noise-induced randomness [30]. |
| QCNN/QRC | Pattern Recognition & Quantum Tasks (e.g., LiH excited energy prediction) | Medium | High (Context-Dependent) | Medium | Amplitude damping can improve performance in shallow circuits (<135 gates, p=0.0005) [13]; Allows for partial adaptation [30]. |
Meta-Heuristic Optimizer Performance for Noisy VQE
For VQE, the choice of classical optimizer is a critical factor in mitigating noise. A large-scale benchmark of over 50 meta-heuristic algorithms revealed significant differences in robustness [80] [99] [100]. The performance of selected optimizers on the 1D Ising and Hubbard models under noisy conditions is quantified below.
Table 2: Optimizer Performance for Noisy VQE Landscapes [80] [99]
| Optimizer | Category | Performance on Noisy VQE | Key Characteristics |
|---|---|---|---|
| CMA-ES | Evolutionary Strategy | Excellent | Consistently top performer; handles rugged, distorted landscapes. |
| iL-SHADE | Evolutionary | Excellent | Robust convergence even with 192 parameters. |
| Simulated Annealing (Cauchy) | Physics-Inspired | Good | Shows robustness to noise. |
| Harmony Search | Music-Inspired | Good | Effective in noisy, multimodal landscapes. |
| Symbiotic Organisms Search | Swarm-Based | Good | Demonstrates reliable performance. |
| PSO, GA, standard DE | Swarm/Evolutionary | Poor | Performance degrades sharply with noise. |
Experimental Protocols for Noise Resilience
Workflow for Systematic Resilience Benchmarking
To empirically evaluate the noise resilience of VQE, QAOA, and QCNN for molecular problems, researchers should adopt a structured experimental workflow. The following diagram outlines the key phases of this process.
Diagram 1: Noise Resilience Benchmarking Workflow
Detailed Methodology for a VQE Resilience Experiment
This section provides a detailed protocol for a VQE noise-injection experiment, based on methodologies used in recent studies [80] [13].
1. Problem Definition and Hamiltonian Formulation:
- Objective: Calculate the ground-state energy of a test molecule, such as Lithium Hydride (LiH) or a hydrogen chain.
- Procedure: Use a classical electronic structure package (e.g., PySCF) to generate the second-quantized molecular Hamiltonian. Map this Hamiltonian to a qubit operator using a transformation like Jordan-Wigner or Bravyi-Kitaev.
2. Ansatz and Circuit Construction:
- Ansatz Selection: Employ a hardware-efficient ansatz suitable for NISQ devices or a chemically inspired ansatz (like UCCSD).
- Parameter Initialization: Initialize parameters using a strategy designed to avoid barren plateaus, such as focusing on the vicinity of the Hartree-Fock state.
3. Noise Model Injection and Simulation:
- Platform: Use a quantum simulator with built-in noise models (e.g., Qiskit AerSimulator or Cirq).
- Noise Configuration: Inject controlled levels of specific noise types after each gate operation. A standard experimental setup would include:
- Depolarizing noise: With error probabilities ( p ) in the range of ( 0.001 ) to ( 0.01 ).
- Amplitude damping: Parameterized by a relaxation rate ( \gamma ), with ( \gamma ) similarly in the range of ( 0.001 ) to ( 0.01 ).
- Phase damping: Also parameterized to match realistic coherence times.
- Measurement: Simulate finite-shot sampling (e.g., 10,000 shots per expectation value estimation) to capture stochastic noise effects.
4. Optimization Loop:
- Optimizer Selection: Based on the benchmarking results in Section 3.2, select a robust optimizer like CMA-ES or iL-SHADE for the noisy setting. For comparison, a standard optimizer like COBYLA or SPSA can be run in parallel.
- Convergence Criterion: Run the optimization until the energy change between iterations falls below a set threshold (e.g., ( 10^{-6} ) Ha) or a maximum number of iterations is reached.
5. Data Collection and Analysis:
- Key Metrics:
- Final Energy Error: Difference between the VQE result and the exact classical result.
- Convergence Speed: Number of iterations or quantum circuit evaluations required.
- Optimization Trace: Plot of energy vs. iteration to visualize landscape traversal.
- Comparative Analysis: Compare the final precision and convergence behavior across different noise models and optimizer pairs.
The Scientist's Toolkit: Research Reagents & Solutions
This section catalogues essential computational tools and strategies, framed as "research reagents," for conducting noise-resilient quantum simulations of molecular problems.
Table 3: Essential Reagents for Noise-Resilient Quantum Chemistry Simulations
| Reagent / Solution | Type | Function in Experiment | Exemplars / Notes |
|---|---|---|---|
| Noise-Resilient Optimizers | Software Algorithm | Navigates distorted, noisy optimization landscapes; avoids local minima. | CMA-ES, iL-SHADE [80] [99]. Prefer over standard PSO or GA. |
| Error Mitigation Framework | Software Protocol | Reduces the impact of noise on circuit outputs without full error correction. | ZNE, PEC, Measurement Error Mitigation [30]. A hybrid APGEM-ZNE-PEC framework is promising for QRL/QAOA. |
| Hardware-Efficient & Noise-Aware Ansatzes | Circuit Architecture | Minimizes circuit depth and aligns circuit symmetries with noise structure to reduce error accumulation. | "Y-axis" rotation ansatzes can be more resilient than "X-axis" for specific noise models [101]. |
| High-Fidelity Quantum Simulators | Software Platform | Emulates NISQ device behavior with configurable noise models for reproducible benchmarking. | Qiskit AerSimulator, Cirq [30]. Allows for controlled noise injection. |
| Metastability Analysis Metric | Analytical Tool | Quantifies the impact of noise that lingers in intermediate states, informing resilient algorithm design. | A practical metric to assess noise resilience without full classical simulation [101]. |
Discussion and Future Directions
The comparative analysis reveals that noise resilience is not a universal property but is highly dependent on the interplay between algorithm architecture, noise type, and classical optimization strategy. VQE and QAOA, while powerful, show high vulnerability to unital noise like depolarizing, which induces barren plateaus. QCNN and related models demonstrate a potential for leveraging structured, non-unital noise like amplitude damping, turning a hardware limitation into a computational asset in specific, constrained scenarios [13] [30].
A critical insight for drug development professionals is that the path to quantum utility in molecular simulation will be incremental. It requires a co-design approach where algorithms are tailored not only to the chemical problem but also to the specific error profile of the target quantum hardware. The findings underscore that simply porting classical quantum chemistry algorithms to quantum hardware is insufficient. Future work must focus on:
- Developing Noise-Aware Ansatzes: Designing parameterized quantum circuits whose inherent structure is naturally resilient to dominant noise channels on a given processor [101].
- Advanced Hybrid Mitigation: Tightly integrating circuit-level error mitigation (like ZNE and PEC) with algorithm-level strategies (like adaptive policy guidance in QRL) to create robust, multi-layered defense systems against noise [30].
- Exploiting Noise as a Feature: Further investigating the conditions under which non-unital noise can be harnessed to improve performance, moving beyond the paradigm of noise as a purely detrimental force [101] [13].
In conclusion, the choice of algorithm for a molecular problem on NISQ devices must be informed by a thorough understanding of its noise resilience profile. For ground-state energy calculations, VQE paired with a robust optimizer like CMA-ES is the current standard, but requires aggressive error mitigation. For other tasks, emerging models like QCNNs may offer superior resilience. As hardware and algorithms co-evolve, this nuanced understanding of noise will be the key to unlocking practical quantum advantages in chemistry and drug discovery.
The pharmaceutical industry is undergoing a profound transformation driven by the convergence of artificial intelligence (AI), quantum computing, and traditional computational chemistry. This technical guide examines the evolving validation frameworks required to bridge small molecule development with complex pharmaceutical compounds, contextualized within quantum noise models for chemistry simulations. As AI-designed small molecules progress through clinical trials and quantum computers advance toward practical chemical simulations, establishing robust, cross-disciplinary validation methodologies becomes paramount for researchers and drug development professionals [102] [103].
Small molecules remain the bedrock of global healthcare, comprising approximately 60% of total pharmaceutical sales with a market value of around $550 billion in 2023 [104]. Despite the emergence of biologics, small molecules offer unique advantages including oral bioavailability, greater stability, lower production costs, and improved tissue penetration into solid tumors [105]. The growing pipeline of AI-designed small molecules exemplifies this trend, with over 75 AI-derived molecules reaching clinical stages by the end of 2024 [102]. Meanwhile, quantum computing promises to overcome the exponential scaling limitations of conventional multi-configurational methods in quantum chemistry, potentially revolutionizing how we simulate molecular systems and predict spectroscopic properties [35].
This guide presents a comprehensive technical framework for validating computational approaches across the drug discovery pipeline, with particular emphasis on addressing quantum noise in chemical simulations—a critical challenge in the noisy intermediate-scale quantum (NISQ) era. We integrate experimental protocols, quantitative comparisons, and visualization tools to equip researchers with practical methodologies for advancing pharmaceutical development.
Small Molecules in the Modern Pharmaceutical Landscape
Strategic Importance and Clinical Advantages
Small molecules continue to demonstrate enduring commercial and therapeutic value despite competition from biologics. Their compact size (typically <1 kDa) enables unique pharmacological advantages, including cell membrane permeability and blood-brain barrier penetration, accessing intracellular and central nervous system targets largely inaccessible to biologics [104]. Oral administration remains a significant clinical advantage, enhancing patient adherence particularly for chronic conditions [104].
The economic case for small molecules remains compelling, with manufacturing costs approximately $5 per pack compared to $60 per pack for biologics [104]. This cost efficiency extends through the development lifecycle, with small-molecule programs often proceeding more quickly and economically through discovery and early development phases despite potentially lower long-term revenue projections [104].
AI-Driven Innovation in Small Molecule Discovery
Artificial intelligence has dramatically accelerated small molecule discovery timelines while enhancing precision. Notable successes include Insilico Medicine's TNIK inhibitor, INS018_055, which progressed from target discovery to Phase II clinical trials in approximately 18 months—a fraction of traditional timelines [103]. Exscientia reported AI-driven design cycles approximately 70% faster requiring 10× fewer synthesized compounds than industry norms [102].
Table 1: AI-Designed Small Molecules in Clinical Development
| Compound | Company | Indication | Phase | Key Results |
|---|---|---|---|---|
| ISM001-055 (rentosertib) | Insilico Medicine | Idiopathic Pulmonary Fibrosis | Phase IIa | Positive Phase IIa results [102] |
| DSP-1181 | Exscientia | Obsessive Compulsive Disorder | Phase I (Discontinued) | First AI-designed drug to enter trials (2020) [102] |
| GTAEXS-617 | Exscientia | Solid Tumors (CDK7 inhibitor) | Phase I/II | Active trial as of 2024 [102] |
| EXS-74539 | Exscientia | LSD1 inhibitor | Phase I | IND approval 2024 [102] |
| Baricitinib | BenevolentAI/Eli Lilly | COVID-19, Rheumatoid Arthritis | Approved/Repurposed | AI-assisted repurposing [103] |
AI technologies deployed across the discovery pipeline include generative models for de novo molecular design, graph neural networks for property prediction, and reinforcement learning for multi-parameter optimization [103] [105]. These approaches complement rather than replace traditional medicinal chemistry, with success heavily dependent on data quality, expert interpretation, and experimental validation [103].
Quantum Computing for Molecular Simulations: Methods and Noise Challenges
Quantum Computational Chemistry Framework
Quantum computational chemistry leverages quantum processing units (QPUs) to overcome exponential scaling limitations in electronic structure calculations. The active space approximation partitions the molecular wavefunction into inactive, active, and virtual components:
|0(θ)〉 = |I〉 ⊗ |A(θ)〉 ⊗ |V〉
where |A(θ)〉 represents the active space prepared on a quantum device using parameterized unitary transformations [35]. This hybrid quantum-classical approach leverages classical computational resources for tractable components while reserving quantum resources for strongly correlated electron interactions.
The trotterized unitary coupled-cluster with singles and doubles (tUCCSD) ansatz provides a common wavefunction parameterization:
[ U(θ) = \prod{k=1}^{N{SD}} \prod{l=1}^{N{Pauli}} e^{iθ{k,l} \hat{P}{k,l}} ]
where (\hat{P}_{k,l}) represents Pauli strings derived from fermionic excitation operators [35]. Orbital optimization introduces additional parameters κ through non-redundant rotations between inactive, active, and virtual spaces.
Quantum Linear Response Theory for Spectroscopic Properties
Quantum linear response (qLR) theory enables the calculation of excited state energies and properties essential for spectroscopic characterization [35]. The generalized eigenvalue problem:
[ E^{[2]} \betak = \omegak S^{[2]} \beta_k ]
yields excitation energies ωk and corresponding excitation vectors βk, where E[2] represents the Hessian matrix and S[2] the metric matrix [35]. This framework extends the capabilities of quantum processors beyond ground-state calculations to dynamic molecular properties.
Quantum Noise Models and Their Impact
Current noisy intermediate-scale quantum (NISQ) devices face significant limitations from various noise sources that affect computational fidelity. Research from Johns Hopkins University has developed advanced frameworks for characterizing how quantum noise propagates through quantum systems using root space decomposition, classifying noise based on how it causes state transitions [38].
Table 2: Quantum Noise Channels and Their Characteristics
| Noise Channel | Type | Mathematical Formulation | Impact on Algorithms |
|---|---|---|---|
| Depolarizing | Unital | (\mathcal{E}(\rho) = (1-p)\rho + p\frac{I}{d}) | Uniformly randomizes quantum state [5] |
| Amplitude Damping | Non-unital (HS-contractive) | (\mathcal{E}(\rho) = \begin{pmatrix} \rho{00} + (1-e^{-T{\text{gate}}/T1})\rho{01} & e^{-T{\text{gate}}/T2}\rho{01}\ e^{-T{\text{gate}}/T2}\rho{10} & e^{-T{\text{gate}}/T1}\rho_{11} \end{pmatrix}) | Energy dissipation to environment [70] [5] |
| Phase Damping | Unital | Diagonal elements preserved, off-diagonal decay | Loss of quantum coherence without energy loss [21] |
| Bit Flip | Unital | (\mathcal{E}(\rho) = (1-p)\rho + p X\rho X) | Classical bit-flip error [21] |
| Phase Flip | Unital | (\mathcal{E}(\rho) = (1-p)\rho + p Z\rho Z) | Classical phase-flip error [21] |
The amplitude damping channel incorporates T1 (energy relaxation time) and T2 (dephasing time) parameters, with the gate error probability derived from the relationship:
[ F{\text{gate}} = (1-p) F{\text{amp-phase}} + \frac{p}{d} ]
where Fgate is the gate fidelity from randomized benchmarking, Famp-phase is the average fidelity of the amplitude phase damping channel, and d is the Hilbert space dimension [70].
Recent research has generalized the study of noise-induced barren plateaus (NIBPs) beyond unital noise to include Hilbert-Schmidt (HS)-contractive non-unital maps like amplitude damping [5]. This work has identified the associated phenomenon of noise-induced limit sets (NILS), where noise pushes the cost function toward a range of values rather than a single value, disrupting variational quantum algorithm training in unexpected ways [5].
Quantum Noise Impact & Mitigation - This diagram illustrates the relationship between quantum noise sources, their algorithmic impacts, and corresponding mitigation strategies.
Integrated Validation Frameworks: Methodologies and Protocols
Experimental Protocol for Quantum Chemical Validation
Validating quantum computational chemistry methods requires systematic comparison against classical benchmarks and experimental data. The following protocol outlines a comprehensive validation approach:
System Selection and Preparation:
- Select benchmark molecular systems with available high-quality experimental spectroscopic data
- Define active spaces encompassing orbitals relevant to target electronic transitions
- Perform classical complete active space self-consistent field (CASSCF) calculations as reference
Quantum Simulation Setup:
- Map fermionic operators to qubit representations using Jordan-Wigner or Bravyi-Kitaev transformations
- Prepare ground state using variational quantum eigensolver (VQE) with tUCCSD ansatz
- Optimize orbital rotation parameters κ and cluster amplitudes θ using gradient-based methods
Quantum Linear Response Implementation:
- Construct Hessian (E[2]) and metric (S[2]) matrices using quantum measurements
- Solve generalized eigenvalue problem on classical co-processor
- Calculate excitation energies and oscillator strengths for spectroscopic properties
Noise Characterization and Mitigation:
- Characterize device-specific noise parameters (T1, T2, gate errors, readout errors)
- Implement zero-noise extrapolation or probabilistic error cancellation
- Apply measurement error mitigation using calibration matrices
Validation Metrics:
- Calculate mean absolute errors for excitation energies relative to CASSCF and experimental values
- Compute spectroscopic fidelity metrics for absorption band shapes and intensities
- Assess computational resource requirements (quantum circuit depth, measurement counts)
AI-Driven Small Molecule Validation Framework
For AI-designed small molecules, comprehensive validation spans computational, experimental, and clinical domains:
Computational Validation:
- Perform molecular dynamics simulations to assess target binding stability
- Calculate binding free energies using advanced sampling methods
- Predict ADMET properties using ensemble machine learning models
- Apply structural alert identification for toxicity risk assessment
Experimental Validation:
- Conduct in vitro binding assays to confirm target engagement
- Assess selectivity profiles against related targets
- Determine pharmacokinetic parameters in relevant model systems
- Evaluate efficacy in disease-relevant cellular models
Clinical Validation:
- Design randomized controlled trials with biomarker-strategic endpoints
- Implement adaptive trial designs for efficient dose optimization
- Incorporate digital health technologies for real-world monitoring
- Collect real-world evidence for post-market safety surveillance
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents and Materials for Validation Studies
| Reagent/Material | Function | Application Context |
|---|---|---|
| Quantum Processing Units | Execution of quantum circuits | Molecular quantum simulations [35] |
| Cryogenic Qubit Control Systems | Qubit initialization, manipulation, readout | NISQ device operation [38] |
| High-Throughput Screening Assays | Compound activity profiling | Hit identification and validation [103] |
| Target-Specific Binding Assays | Confirm target engagement and potency | Mechanism of action studies [105] |
| Cellular Disease Models | Efficacy assessment in biologically relevant systems | Preclinical validation [105] |
| Multi-omics Profiling Platforms | Comprehensive molecular characterization | Patient stratification biomarker discovery [105] |
| ADMET Prediction Software | Computational pharmacokinetic and toxicity screening | Lead optimization [103] |
Regulatory and Implementation Considerations
Evolving Regulatory Landscape for AI and Quantum-Enabled Drug Development
Regulatory agencies are adapting to the emergence of AI and advanced computational methods in pharmaceutical development. The FDA's draft guidance "Considerations for the Use of Artificial Intelligence to Support Regulatory Decision Making for Drug and Biological Products" (2025) establishes a risk-based credibility assessment framework informed by over 500 submissions with AI components from 2016 to 2023 [103]. The FDA Modernization Act 3.0 formally recognizes human-relevant alternative models—including organ-on-chip systems, computational modeling, and AI-driven in silico approaches—as viable substitutes for traditional animal testing [105].
The INFORMED (Information Exchange and Data Transformation) initiative at the FDA from 2015-2019 functioned as a multidisciplinary incubator for deploying advanced analytics across regulatory functions, creating a template for regulatory innovation [106]. This initiative demonstrated the value of protected experimentation spaces within regulatory agencies and highlighted the importance of multidisciplinary teams integrating clinical, technical, and regulatory expertise.
Implementation Challenges and Strategic Recommendations
Successful implementation of integrated validation frameworks faces several challenges:
Data Quality and Standardization: Inconsistent data quality across sources impedes model training and validation. Recommendation: Establish standardized data collection protocols and implement rigorous quality control measures.
Algorithmic Transparency: Limited interpretability of complex AI and quantum algorithms hinders regulatory acceptance. Recommendation: Develop explainability frameworks and uncertainty quantification methods.
Computational Resource Requirements: Quantum chemical simulations and large-scale AI training demand substantial computational resources. Recommendation: Implement hybrid cloud computing strategies and optimize algorithmic efficiency.
Cross-Disciplinary Knowledge Gaps: Specialized expertise in both pharmaceutical development and advanced computing remains rare. Recommendation: Foster interdisciplinary training programs and collaborative research networks.
Regulatory Alignment: Evolving regulatory standards create uncertainty for technology developers. Recommendation: Engage early with regulatory agencies through pre-submission meetings and pilot programs.
Validation Framework Workflow - This diagram outlines the integrated validation framework from data inputs through computational methods to validated outputs.
The convergence of AI, quantum computing, and traditional pharmaceutical science demands robust, integrated validation frameworks that span computational predictions to clinical outcomes. As quantum hardware advances beyond the NISQ era, noise-resilient algorithms will enable increasingly accurate molecular simulations, potentially revolutionizing early drug discovery. Similarly, AI methodologies are evolving from pattern recognition tools to autonomous discovery systems, with agentic AI beginning to navigate entire discovery pipelines [103].
Future validation frameworks must address several critical frontiers. First, standardized benchmarking datasets and protocols for quantum chemical calculations will enable meaningful comparison across platforms and algorithms. Second, prospective clinical validation of AI-designed compounds must move beyond proof-of-concept to demonstrate impact on patient outcomes. Third, regulatory science must continue evolving to accommodate the unique characteristics of AI and quantum-enabled drug development while maintaining rigorous safety standards.
The most successful pharmaceutical innovators will be those who strategically integrate these advanced computational technologies within a rigorous validation framework that respects the complementary strengths of traditional and emerging approaches. By establishing robust methodologies for validating predictions from initial quantum simulations through clinical outcomes, researchers can accelerate the development of safer, more effective therapeutics while managing the inherent uncertainties of innovative technologies.
Cross-Platform Performance Evaluation on Different Quantum Hardware
The pursuit of practical quantum advantage, particularly in chemistry simulations and drug development, hinges on the ability to accurately evaluate and predict the performance of quantum algorithms across diverse hardware platforms. The current quantum computing landscape is characterized by a fragmentation of hardware technologies and architectures, making objective, cross-platform performance assessment both challenging and essential [107]. For researchers in fields like pharmaceutical development, where quantum simulations of molecules such as Cytochrome P450 promise to accelerate drug discovery timelines, understanding how different hardware platforms handle realistic noise is critical for selecting appropriate quantum resources [68].
This technical guide provides a comprehensive framework for evaluating quantum hardware performance specifically within the context of quantum noise models relevant to chemistry simulations. We focus particularly on depolarizing and amplitude damping noise—two critical noise processes that significantly impact the fidelity of quantum chemical calculations—and present standardized methodologies for cross-platform comparison that account for the complex interplay between hardware capabilities, error characteristics, and algorithmic requirements.
Theoretical Foundations: Quantum Noise Models for Chemistry Simulations
Mathematical Framework of Quantum Noise Channels
In real quantum devices, noise originates from various factors including imperfect quantum operations and environmental interactions. These effects are mathematically described using the language of quantum channels and density matrices, which generalize the concept of quantum states and their evolution beyond ideal unitary transformations [108] [109].
Quantum channels are represented as completely positive, trace-preserving (CPTP) maps that transform an initial state Ï according to:
[\varepsilon(\rho) =\sum{k=0}^{m-1} E{k} \rho E_{k}^{\dagger}]
where ({Ek}) are Kraus operators satisfying the completeness condition (\sumk Ek^\dagger Ek = I) [109]. This formalism captures the statistical nature of noisy quantum evolution, where the output state represents a probability distribution over possible transformations.
Key Noise Models in Chemical Simulation
For chemistry simulations, particularly those targeting molecular energy calculations using algorithms like Variational Quantum Eigensolver (VQE) or Quantum Phase Estimation (QPE), two specific noise models are particularly relevant:
Depolarizing Channel: This model represents the gradual loss of quantum information to the environment, described by:
[\varepsilon_{DF}( \rho ) =(1-p)\rho + \frac{p}{4}(I \rho I + X \rho X+ Y \rho Y+Z \rho Z)]
with Kraus operators (K0 = \sqrt{1-p}I), (K1 = \sqrt{p/3}X), (K2 = \sqrt{p/3}Y), (K3 = \sqrt{p/3}Z) [108] [109]. In chemical simulations, depolarizing noise leads to the gradual destruction of carefully prepared entangled states essential for representing molecular wavefunctions.
Amplitude Damping Channel: This model describes energy dissipation, crucial for modeling molecular relaxation processes, with Kraus operators:
[E{0} =\begin{bmatrix} 1 & 0\ 0 & \sqrt{1-\gamma } \end{bmatrix}, E{1} =\begin{bmatrix} 0 & \sqrt{\gamma }\ 0 & 0 \end{bmatrix}]
The amplitude damping channel causes qubits to decay from excited state (|1\rangle) to ground state (|0\rangle) with probability γ [109]. For molecular simulations, this represents both a source of error and a physically relevant process when studying relaxation dynamics.
Table 1: Quantum Noise Channels Relevant to Chemistry Simulations
| Noise Channel | Kraus Operators | Physical Process | Impact on Chemistry Simulations |
|---|---|---|---|
| Depolarizing | (K0 = \sqrt{1-p}I), (K1 = \sqrt{p/3}X),(K2 = \sqrt{p/3}Y), (K3 = \sqrt{p/3}Z) | Unstructured information loss | Destroys entanglement in molecular wavefunctions |
| Amplitude Damping | (E{0} =\begin{bmatrix} 1 & 0\ 0 & \sqrt{1-\gamma } \end{bmatrix}),(E{1} =\begin{bmatrix} 0 & \sqrt{\gamma }\ 0 & 0 \end{bmatrix}) | Energy dissipation | Models molecular relaxation; causes computational errors |
| Phase Damping | (E{0} =\begin{bmatrix} 1 & 0\ 0 & \sqrt{1-\gamma } \end{bmatrix}),(E{1} =\begin{bmatrix} 0 & 0\ 0 & \sqrt{\gamma } \end{bmatrix}) | Pure dephasing | Destroys phase coherence in superposition states |
| Bit Flip | (E0=\sqrt{1-p}I), (E{1} =\sqrt{p}X) | Classical bit errors | Introduces population errors in computational basis |
Current Quantum Hardware Landscape and Benchmarking Initiatives
Hardware Performance Breakthroughs in 2025
The quantum hardware landscape has witnessed remarkable advances in 2025, with particularly significant progress in error correction capabilities. Google's Willow quantum chip, featuring 105 superconducting qubits, demonstrated exponential error reduction as qubit counts increased—a critical milestone known as going "below threshold" [68]. The Willow chip completed a benchmark calculation in approximately five minutes that would require a classical supercomputer 10^25 years to perform, providing strong evidence that large, error-corrected quantum computers are feasible.
IBM has unveiled its fault-tolerant roadmap centered on the Quantum Starling system targeted for 2029, which will feature 200 logical qubits capable of executing 100 million error-corrected operations [68]. Meanwhile, Microsoft introduced Majorana 1, a topological qubit architecture built on novel superconducting materials designed to achieve inherent stability with reduced error correction overhead. These advances have pushed error rates to record lows of 0.000015% per operation, with researchers achieving coherence times of up to 0.6 milliseconds for the best-performing qubits [68].
Standardized Benchmarking Frameworks
The fragmentation of quantum hardware technologies has created an urgent need for standardized evaluation methods. In response, QunaSys released "QURI Bench," a benchmark report specifically designed to provide objective, cross-platform evaluation of quantum hardware performance for chemistry and materials science applications [107].
This benchmark focuses on two specific systems highly relevant to pharmaceutical research: p-benzyne (a reactive intermediate in organic chemistry) and the 2D Fermi-Hubbard model (a fundamental model for strongly correlated electron systems) [107]. Rather than using the resource-intensive Quantum Phase Estimation (QPE) algorithm, the benchmark employs Gaussian Statistical Phase Estimation (SPE) as a more realistic near-term algorithm, acknowledging current hardware limitations.
Table 2: Quantum Hardware Performance Benchmarks for Chemistry Applications (2025)
| Hardware Platform | Qubit Count | Error Rate/Operation | Max Active Orbitals (p-benzyne) | Max Lattice Size (Fermi-Hubbard) | Coherence Time |
|---|---|---|---|---|---|
| Google Willow | 105 qubits | Not specified | 26 orbitals | 10x10 lattice | Not specified |
| IBM Quantum Starling | 200 logical (planned) | 100 million error-corrected ops | Not specified | Not specified | Not specified |
| Microsoft Majorana 1 | 28 logical/112 physical | 1,000-fold error reduction | Not specified | Not specified | Not specified |
| IonQ | 36 qubits | Not specified | Not specified | Not specified | Not specified |
The benchmarking methodology makes several key assumptions that reflect current hardware constraints: devices must have sufficient logical qubits with specified error rates in roadmaps; executable gate counts must exceed SPE implementation requirements; Clifford + T decomposition is assumed; and parallelization of gate execution is not considered [107]. This creates a standardized framework for evaluating hardware readiness for specific chemical applications.
Experimental Protocols for Cross-Platform Evaluation
Standardized Evaluation Workflow
The evaluation of quantum hardware for chemistry applications requires a systematic approach that accounts for both algorithmic requirements and hardware characteristics. The following workflow diagram illustrates the standardized evaluation process:
Diagram 1: Hardware Evaluation Workflow
Resource Estimation Methodology
Accurate resource estimation is fundamental to predicting hardware performance for specific chemical problems. The QURI Bench methodology provides a structured approach to this estimation [107]:
Logical Qubit Requirements: Determine the number of logical qubits needed to represent the target chemical system. For p-benzyne, active space sizes of [6, 14, 18, 26] orbitals are computed, with each orbital typically requiring one logical qubit.
Gate Count Estimation: Calculate the total number of gates required to implement the target algorithm (SPE in this case), assuming Clifford + T decomposition.
Error Budget Allocation: A device is considered capable of executing SPE when the number of gates it can execute exceeds the algorithm requirements, assuming a 33% logical error is acceptable after N gate executions.
Hardware Capability Assessment: Compare resource requirements against hardware roadmaps, considering both qubit counts and error rates.
This methodology explicitly does not consider performance improvements from error mitigation techniques or parallelization of gate execution, providing a conservative estimate of hardware capabilities [107].
Noise-Aware Circuit Compilation
The compilation of quantum circuits for chemical simulations must account for specific hardware constraints and noise characteristics. The following diagram illustrates the noise-aware compilation process:
Diagram 2: Noise-Aware Circuit Compilation
For nuclear magnetic resonance (NMR) based systems like the SPINQ Gemini Lab, pulse-level control enables direct manipulation of quantum states through RF pulses, providing a unique opportunity to study noise processes at the fundamental level [110]. This hands-on access to pulse control allows researchers to develop deeper intuition about how noise affects quantum computations.
Case Study: Quantum Hardware Evaluation for Pharmaceutical Applications
Documented Quantum Advantage in Pharmaceutical Simulations
Recent advances have demonstrated tangible progress toward practical quantum advantage in pharmaceutical research. In March 2025, IonQ and Ansys achieved a significant milestone by running a medical device simulation on IonQ's 36-qubit computer that outperformed classical high-performance computing by 12 percent—one of the first documented cases of quantum computing delivering practical advantage over classical methods in a real-world application [68].
Similarly, Google's collaboration with Boehringer Ingelheim demonstrated quantum simulation of Cytochrome P450, a key human enzyme involved in drug metabolism, with greater efficiency and precision than traditional methods [68]. These advances could significantly accelerate drug development timelines and improve predictions of drug interactions and treatment efficacy.
Performance Metrics for Chemistry Simulations
When evaluating quantum hardware for chemical simulations, several key metrics provide insight into platform suitability:
Algorithmic Fidelity: The accuracy with which the target algorithm executes, typically measured by comparing results against classical benchmarks or theoretical values.
Resource Scaling: How qubit counts, gate counts, and execution times scale with problem size (e.g., number of orbitals in molecular simulations).
Noise Resilience: The algorithm's performance degradation under realistic noise models, particularly depolarizing and amplitude damping noise.
Time-to-Solution: The total computational time required to achieve chemical accuracy (typically 1 kcal/mol for molecular energies).
The National Energy Research Scientific Computing Center has conducted studies indicating that quantum resource requirements have declined sharply while industry roadmaps project hardware capabilities rising steeply [68]. Their analysis suggests that quantum systems could address Department of Energy scientific workloads—including materials science, quantum chemistry, and high-energy physics—within five to ten years.
Table 3: Research Reagent Solutions for Quantum Chemistry Experiments
| Tool/Resource | Function | Example Implementation |
|---|---|---|
| Quantum Hardware Simulators | Simulate ideal and noisy quantum circuits | PennyLane default.mixed device [108], MindSpore Quantum [109] |
| Noise Channel Libraries | Implement depolarizing, amplitude damping, and other noise models | PennyLane's BitFlip, DepolarizingChannel, AmplitudeDamping [108] |
| Chemical System Encoders | Map molecular systems to qubit representations | QURI Bench p-benzyne and Fermi-Hubbard models [107] |
| Benchmarking Suites | Standardized performance evaluation across platforms | QURI Bench [107] |
| Educational Platforms | Hands-on experimentation with real quantum systems | SPINQ Gemini Lab [110] |
| Virtual Laboratories | Prototype experimental setups and visualize quantum states | Quantum Flytrap Virtual Lab [111] |
| Error Mitigation Tools | Reduce impact of noise on computational results | Ignored in conservative benchmarks but essential in practice [107] |
The cross-platform evaluation of quantum hardware for chemistry simulations requires a nuanced approach that balances theoretical algorithmic requirements with practical hardware constraints. As demonstrated by recent benchmarks and documented cases of quantum advantage in pharmaceutical applications, the field is progressing rapidly toward practical utility.
For researchers in drug development and chemistry simulations, successful navigation of this landscape requires understanding both the specific noise processes affecting their calculations—particularly depolarizing and amplitude damping noise—and the standardized methodologies for evaluating hardware against these challenges. The tools, frameworks, and experimental protocols outlined in this guide provide a foundation for making informed decisions about quantum hardware selection and investment as the technology continues to mature toward broader practical application.
Statistical Validation Methods and Confidence Metrics for Drug Discovery Applications
In the contemporary pharmaceutical landscape, statistical validation provides the critical framework for ensuring that drug discovery methods yield reliable, reproducible, and meaningful results. The transition towards high-throughput technologies and artificial intelligence (AI) has intensified the need for robust statistical frameworks that can quantify confidence and manage uncertainty [112] [113]. These methodologies are paramount for making informed go/no-go decisions, prioritizing resource allocation, and ultimately reducing the high attrition rates that have long plagued the drug development pipeline. Furthermore, in the context of emerging computational paradigms like quantum computing for chemistry simulations, understanding and mitigating the impact of quantum noise models—such as depolarizing and amplitude damping—becomes essential for validating any resultant predictions [30]. This guide details the core statistical validation methods and confidence metrics that are foundational to modern drug discovery, providing researchers with the tools to critically evaluate experimental and computational outcomes across diverse modalities.
The evolution of drug discovery has been significantly influenced by Eroom's Law (the inverse of Moore's Law), which observes that the cost of developing a new drug has steadily increased over time despite technological advancements [112]. This paradox underscores the critical need for more efficient and predictive R&D strategies. Statistical validation acts as a counterweight to this trend by introducing rigor, predictability, and quantitative risk assessment into every stage of the process, from initial target identification to late-stage lead optimization [113] [114]. By embedding robust statistical principles into the discovery workflow, organizations can enhance translational predictivity, compress development timelines, and increase the probability of technical and regulatory success.
Core Statistical Validation Frameworks
Model-Informed Drug Development (MIDD)
Model-Informed Drug Development (MIDD) has emerged as a cornerstone of modern pharmaceutical R&D, providing a quantitative, data-driven framework for decision-making from discovery through post-market surveillance. The core principle of MIDD is a "fit-for-purpose" strategy, which mandates that the selected modeling and simulation tools must be closely aligned with the Key Questions of Interest (QOI) and the specific Context of Use (COU) [114]. This approach ensures that models are neither overly simplistic nor unnecessarily complex for the decision at hand. MIDD's utility is recognized by global regulatory agencies, and efforts by the International Council for Harmonisation (ICH), such as the M15 guidance, aim to standardize its application across international borders, thereby promoting consistency and efficiency in drug development [114].
The following table summarizes the primary quantitative tools utilized within the MIDD framework and their specific applications in the drug development lifecycle.
| Tool/Methodology | Primary Application in Drug Discovery & Development |
|---|---|
| Quantitative Structure-Activity Relationship (QSAR) | Predicts biological activity and ADMET properties based on a compound's chemical structure [114]. |
| Physiologically Based Pharmacokinetic (PBPK) | Mechanistically simulates drug absorption, distribution, metabolism, and excretion (ADME) [114]. |
| Population PK/Exposure-Response (ER) | Characterizes variability in drug exposure and its relationship to efficacy and safety outcomes in a population [114]. |
| Quantitative Systems Pharmacology (QSP) | Integrates systems biology and pharmacology to generate mechanism-based predictions of drug behavior and treatment effects [114]. |
| AI/ML Models | Analyzes large-scale biological and chemical datasets to predict targets, optimize compounds, and forecast ADMET properties [114]. |
| Uncertainty Quantification (UQ) | Estimates confidence in predictions, which is critical for resource allocation and risk assessment [115]. |
Uncertainty Quantification (UQ) in Machine Learning
The proliferation of machine learning (ML) in drug discovery has made Uncertainty Quantification (UQ) an indispensable statistical discipline. Accurate UQ allows researchers to discern not just what a model predicts, but how confident that prediction is, which is vital when deciding which virtual compounds to synthesize and test in costly experimental assays [115]. Standard UQ methods include ensemble techniques (where multiple models are trained to assess prediction variance), Bayesian models (which provide a probabilistic framework for uncertainty), and Gaussian processes [115]. A significant advancement in this field is the adaptation of these models to learn from censored regression labels. In pharmaceutical assays, it is common for experimental readings to fall outside a quantifiable range, resulting in censored data (e.g., ">10μM" for low-potency compounds). Traditional ML models discard or mishandle this information. By integrating the Tobit model from survival analysis, ensemble, Bayesian, and Gaussian models can now leverage this partial information, leading to more reliable uncertainty estimates, especially in real-world settings where over 30% of experimental data may be censored [115].
Lifecycle Management and Risk-Based Validation
A paradigm shift is occurring in analytical science, moving from a one-time validation event to a comprehensive lifecycle management approach for analytical methods, as championed by ICH guidelines Q2(R2) and Q14 [116]. This lifecycle strategy encompasses three continuous stages: (1) method design and feasibility studies using Quality-by-Design (QbD) principles, (2) method qualification and validation, and (3) ongoing method performance monitoring during routine use [116]. Central to this framework is the application of risk-based validation. Methodologies like Failure Modes and Effects Analysis (FMEA) are employed to identify and prioritize potential factors that could impact method performance [117]. This allows teams to focus their validation efforts and resources on the most critical parameters—such as specificity, accuracy, precision, and robustness for chromatographic methods—that directly impact product quality and patient safety, thereby optimizing both compliance and operational efficiency [116] [117].
Confidence Metrics for Key Drug Discovery Applications
Target Identification and Validation
In the initial phase of discovery, confidence in a therapeutic target is paramount. The rise of large-scale, multi-omics datasets has enabled the development of computational tools that provide quantitative confidence metrics for target prediction. For instance, the DeepTarget tool leverages drug and genetic knockdown viability screens, integrated with omics data, to predict both primary and secondary (off-target) effects of small molecules [118]. The confidence metric for its predictions is benchmarked through rigorous testing on high-confidence drug-target pairs. In one published study, DeepTarget demonstrated superior performance by accurately predicting mutation-specific drug responses, such as the influence of EGFR T790 mutations on ibrutinib response in BTK-negative solid tumors [118]. The tool's ability to mirror real-world drug mechanisms, where cellular context and pathway-level effects are crucial, provides a more holistic confidence score compared to methods focused solely on direct binding interactions [118].
"Fit-for-Purpose" Analytical Method Validation
The validation of analytical methods used to quantify drug substances and products must be "fit-for-purpose," with confidence metrics tailored to the method's stage and application. The following table outlines the core validation parameters and the statistical measures used to establish confidence for each.
| Validation Parameter | Statistical Confidence Metrics & Methodology |
|---|---|
| Accuracy | Percent recovery of known amounts of analyte; reported as mean % recovery and confidence intervals (e.g., 95% CI). |
| Precision |
|
| Specificity | Demonstration that the method can unequivocally assess the analyte in the presence of potential interferents (e.g., impurities). |
| Linearity & Range | Correlation coefficient (R²), y-intercept, and residual sum of squares from a linear regression model across the specified range. |
| Robustness | Statistical analysis (e.g., ANOVA, Plackett-Burman design) to evaluate the method's resilience to deliberate, small parameter fluctuations. |
These parameters are evaluated under stringent guidelines like ICH Q2(R2), with acceptance criteria pre-defined based on the method's risk and context of use [116]. The trend is towards a lifecycle approach, where validation is not a single event but involves continuous performance monitoring using statistical process control (SPC) charts to track metrics like system suitability test results over time, ensuring ongoing method robustness [116].
AI and Computational Model Validation
Validating AI and computational models requires a distinct set of confidence metrics that go beyond simple goodness-of-fit measures. Prospective, blind challenge trials are considered the gold standard for establishing model credibility. In this paradigm, models are evaluated on data they have never seen before, with the ground truth revealed only after predictions are submitted [119]. This approach was instrumental in validating breakthrough technologies like AlphaFold for protein structure prediction. Key performance metrics in these challenges include:
- Approximation Ratio: How close a solution is to the known optimum, commonly used in optimization tasks like molecular docking [30].
- Fidelity: The similarity between a predicted state and the true state, often used in quantum simulations [30].
- Area Under the Curve (AUC): For classification tasks (e.g., active/inactive), the AUC of the Receiver Operating Characteristic (ROC) curve measures predictive power.
- Calibration Plots: These assess if a model's predicted probabilities of an event (e.g., 80% chance of activity) match the observed frequencies (e.g., activity is indeed observed in 8 out of 10 compounds with an 80% prediction) [115].
Furthermore, defining a model's applicability domain is a critical confidence metric. It statistically defines the chemical or biological space where the model's predictions are reliable, helping researchers avoid extrapolation errors [119].
Advanced Protocols and Workflows
Protocol for Uncertainty Quantification with Censored Data
Objective: To reliably quantify uncertainty in regression models for molecular property prediction (e.g., ICâ‚…â‚€) using both precise and censored experimental labels.
Materials & Reagents:
- Dataset: Containing precise values and censored labels (e.g.,
>10µM). - Software Environment: Python with PyTorch/TensorFlow, and specialized libraries for survival analysis.
- Computational Resources: GPUs are recommended for training deep learning models.
Methodology:
- Data Preprocessing: Separate data into three sets: training, validation (for hyperparameter tuning), and a temporal test set (compounds selected after the model's training data was generated) to simulate real-world performance decay [115].
- Model Integration with Tobit Likelihood: Adapt standard regression models (Ensemble, Bayesian Neural Networks, etc.).
- For precise measurements, use a standard loss function (e.g., Mean Squared Error).
- For censored data (e.g.,
y_censored > threshold), replace the loss with the Tobit likelihood, which models the probability of the data being censored.
- Model Training: Train the adapted model on the combined dataset (precise and censored data).
- Uncertainty Estimation: Generate predictions and uncertainty intervals (e.g., 95% prediction intervals) from the trained model.
- Validation & Evaluation:
- On Non-Censored Test Data: Use calibration plots to check if the predicted uncertainties match the observed errors (e.g., for 95% prediction intervals, ~95% of the true values should fall within the interval).
- Comparative Analysis: Benchmark the model trained with censored data against a baseline model trained only on precise data, demonstrating improved uncertainty estimation on the temporal test set [115].
Protocol for a "Fit-for-Purpose" Analytical Method Lifecycle
Objective: To develop and validate an analytical method following a risk-based lifecycle approach per ICH Q14.
Materials & Reagents:
- Analytical Instrumentation (e.g., UHPLC, HRMS).
- Reference Standards (analyte and potential impurities).
- Data Integrity-Compliant Software (e.g., a LIMS that follows ALCOA+ principles).
Methodology:
- Method Design (QbD Phase):
- Define the Analytical Target Profile (ATP), which specifies the method's required performance.
- Use Risk Assessment (e.g., Ishikawa diagram) to identify critical method parameters.
- Employ Design of Experiments (DoE) to systematically explore the parameter space (e.g., pH, temperature, gradient) and establish a Method Operational Design Range (MODR) that ensures robustness [116].
- Method Qualification:
- Execute protocols to evaluate the validation parameters (Accuracy, Precision, etc.) listed in Section 3.2, using pre-defined acceptance criteria.
- Perform robustness testing within the MODR established from the DoE.
- Ongoing Lifecycle Management:
- Implement Continuous Process Verification by routinely monitoring system suitability tests and quality control sample results.
- Use Statistical Process Control (SPC) charts to track method performance over time. Establish control limits and investigate any out-of-trend (OOT) or out-of-specification (OOS) results.
- Manage method changes through a structured protocol, requiring re-validation only for modifications that impact critical parameters [116] [117].
The following workflow diagram illustrates the key stages of this lifecycle management process.
Diagram 1: Analytical Method Lifecycle Workflow. This chart visualizes the "fit-for-purpose" lifecycle approach, from initial design based on an Analytical Target Profile (ATP) through continuous monitoring and iterative improvement.
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table details key reagents, computational tools, and databases essential for implementing robust statistical validation in drug discovery.
| Tool / Reagent / Database | Type | Function in Validation |
|---|---|---|
| HCDT 2.0 Database | Database | Provides a comprehensive, high-confidence set of drug-target interactions (drug-gene, drug-RNA, drug-pathway) for benchmarking computational prediction tools [120]. |
| CETSA (Cellular Thermal Shift Assay) | Experimental Assay | Provides direct, physiologically relevant evidence of target engagement in intact cells, validating computational predictions of binding [113]. |
| DeepTarget | Computational Tool | Predicts primary and secondary drug targets; its predictions are validated through benchmark testing on known drug-target pairs and experimental case studies [118]. |
| OpenADMET Data & Challenges | Data & Benchmarking Platform | Provides high-quality, consistently generated experimental ADMET data used to train, test, and validate ML models through blind challenges [119]. |
| PBPK/QSAR/QSP Models | Computational Model | MIDD tools used for predictive simulation of drug pharmacokinetics, activity, and system-level effects; validated against in vitro and in vivo data [114]. |
| Tobit Model Integration | Statistical Method | Enhances uncertainty quantification for ML models by allowing them to learn from censored experimental data, providing more reliable confidence intervals [115]. |
The Interface with Quantum Noise Simulation Research
The application of quantum computing to drug discovery, particularly for molecular simulations, introduces a new dimension of statistical validation: resilience to quantum noise. Current Noisy Intermediate-Scale Quantum (NISQ) devices are affected by various noise models, including depolarizing noise (which randomizes the quantum state), amplitude damping (energy dissipation), and dephasing (loss of phase information) [30]. The confidence in any simulation run on such hardware depends on mitigating these effects. Research in this domain focuses on developing error mitigation frameworks that are themselves validated using classical metrics.
For example, a proposed hybrid framework combines Zero-Noise Extrapolation (ZNE), Probabilistic Error Cancellation (PEC), and Adaptive Policy-Guided Error Mitigation (APGEM) [30]. The performance and, consequently, the confidence in quantum simulations are quantified by benchmarking the mitigated results against known classical solutions or synthetic benchmarks. Key validation metrics in this context include the approximation ratio (closeness to the optimal solution), quantum state fidelity (similarity to the ideal state), and learning entropy (measuring the stability of the learning process) [30]. As illustrated in the diagram below, integrating these mitigation strategies is essential for deriving reliable results from quantum computations, drawing a parallel to the use of uncertainty quantification in classical AI/ML models.
Diagram 2: Quantum Noise Mitigation & Validation Workflow. This chart outlines the process of running a quantum simulation, applying a hybrid error mitigation framework to counteract specific noise models, and finally validating the output using standardized confidence metrics.
Statistical validation methods and confidence metrics are the bedrock of efficient and successful modern drug discovery. The field is moving towards more dynamic, integrated, and lifecycle-oriented frameworks such as MIDD, QbD, and continuous process validation. The ability to accurately quantify uncertainty, whether in classical AI models dealing with censored data or in quantum simulations contending with hardware noise, is no longer a luxury but a necessity for making data-driven decisions. As technologies continue to evolve, the commitment to robust, "fit-for-purpose" statistical rigor will be the key differentiator in translating innovative scientific discoveries into safe and effective therapies for patients.
The accurate simulation of molecular systems represents one of the most promising applications of quantum computing, with potential to revolutionize drug discovery, materials science, and chemical engineering. However, the path to practical quantum advantage in chemistry simulations is paved with significant challenges, primarily stemming from the inherent noise in contemporary quantum hardware. This technical guide provides an in-depth examination of how noise affects quantum chemistry simulations, comparing idealized theoretical models, noisy simulations, and results from actual quantum hardware for benchmark molecular systems. Framed within broader research on quantum noise models—particularly depolarizing and amplitude damping channels—this analysis offers researchers and drug development professionals a comprehensive framework for evaluating the current state and near-term potential of quantum computational chemistry.
The NISQ (Noisy Intermediate-Scale Quantum) era is characterized by quantum processors containing dozens to hundreds of qubits with error rates that significantly impact computational fidelity [92] [121]. Understanding the behavior of quantum algorithms under these realistic conditions is essential for advancing the field toward practical applications. This case study focuses specifically on variational quantum eigensolver (VQE) applications to molecular systems, as this hybrid quantum-classical algorithm has emerged as a leading candidate for near-term quantum chemistry computations [122].
Theoretical Framework: Quantum Noise Models for Chemistry Simulations
Mathematical Foundation of Quantum Noise Channels
In quantum information theory, noise processes are formally described using the language of quantum channels and Kraus operators. The evolution of a quantum state Ï under a noisy channel ε can be represented as:
[\varepsilon(\rho) = \sum{k=0}^{m-1} Ek \rho E_k^{\dagger}]
where ({Ek}) are Kraus operators satisfying the completeness condition (\sumk Ek^\dagger Ek = I) [109] [26]. This mathematical framework provides the foundation for modeling various types of noise encountered in quantum hardware.
Classification of Quantum Noise Channels
Table 1: Classification of Common Quantum Noise Channels in Chemical Simulations
| Noise Type | Mathematical Representation | Physical Interpretation | Impact on Chemistry Simulations | ||
|---|---|---|---|---|---|
| Depolarizing Channel | (\varepsilon_{DF}(\rho) = (1-p)\rho + \frac{p}{4}(I\rho I + X\rho X + Y\rho Y + Z\rho Z)) [109] | With probability p, the qubit is replaced by a completely mixed state | Introduces uniform errors across all quantum states, degrading measurement outcomes | ||
| Amplitude Damping Channel | (\varepsilon{AD}(\rho) = E0\rho E0^\dagger + E1\rho E1^\dagger) with (E0 = \begin{bmatrix} 1 & 0 \ 0 & \sqrt{1-\gamma} \end{bmatrix}), (E_1 = \begin{bmatrix} 0 & \sqrt{\gamma} \ 0 & 0 \end{bmatrix}) [109] | Models energy dissipation from excited | (\rangle) to ground | (\rangle) state | Particularly relevant for molecular excited state calculations |
| Phase Damping Channel | (\varepsilon{PD}(\rho) = E0\rho E0^\dagger + E1\rho E1^\dagger) with (E0 = \begin{bmatrix} 1 & 0 \ 0 & \sqrt{1-\gamma} \end{bmatrix}), (E_1 = \begin{bmatrix} 0 & 0 \ 0 & \sqrt{\gamma} \end{bmatrix}) [109] | Models loss of quantum information without energy loss | Degrades coherence and entanglement crucial for quantum algorithms | ||
| Bit Flip Channel | (\varepsilon_{BF}(\rho) = (1-p)I\rho I + pX\rho X) [26] | Qubit state flips | (\rangle \leftrightarrow | 1\rangle) with probability p | Introduces computational basis errors in measurements |
Noise-Aware Quantum Circuit Simulation
The simulation of noisy quantum circuits requires specialized approaches that account for non-unitary evolution. As illustrated in the MindSpore Quantum and Paddle Quantum frameworks, two primary methods exist for this purpose: the density matrix approach, which directly models the evolution of mixed states, and the Monte Carlo wavefunction approach, which uses repeated sampling of possible noise trajectories [109] [26]. The latter approach, while computationally efficient for large systems, introduces statistical uncertainty that must be accounted for in precision-critical applications like molecular energy calculations.
Methodology: Experimental Protocols for Comparative Analysis
Benchmark Molecular Systems Selection
For this comparative analysis, we focus on small aluminum clusters (Alâ», Alâ‚‚, Al₃â») as referenced in the BenchQC study [122]. These systems provide an ideal testbed for multiple reasons: their intermediate complexity bridges the gap between simple diatomic molecules and more complex systems, they exhibit strong electron correlation effects that challenge classical methods, and their well-characterized properties enable reliable benchmarking against established computational and experimental results.
Quantum Simulation Workflow
The standardized workflow for quantum chemistry simulations follows a multi-stage process that integrates both classical and quantum computational resources, as implemented in the BenchQC framework [122]:
Figure 1: Quantum-DFT Embedding Workflow for Molecular Energy Calculations
Research Reagent Solutions: Essential Tools for Quantum Chemistry
Table 2: Essential Software and Hardware Tools for Quantum Chemistry Experiments
| Tool Category | Specific Examples | Function in Chemistry Workflow | Relevant Citation |
|---|---|---|---|
| Quantum SDKs | Qiskit, Paddle Quantum, MindSpore Quantum | Provides noise model simulation, circuit construction, and algorithm implementation | [122] [26] [109] |
| Classical Chemistry Packages | PySCF | Performs preliminary molecular orbital calculations and active space selection | [122] |
| Hardware Platforms | IQM 20-qubit superconducting processor, Quantinuum H2 trapped-ion system | Provides physical quantum devices for experimental validation | [121] [94] |
| Error Mitigation Tools | Zero-noise extrapolation, probabilistic error cancellation | Reduces impact of noise on measurement outcomes | [13] |
| Benchmarking Frameworks | BenchQC | Standardizes comparison across different computational approaches | [122] |
Case Study: Aluminum Clusters Quantum Simulation
Experimental Parameters and Configurations
The BenchQC study implemented a comprehensive benchmarking approach for aluminum clusters, systematically varying key parameters to assess their impact on simulation accuracy [122]:
- Classical Optimizers: SLSQP, COBYLA, L-BFGS-B
- Circuit Types: EfficientSU2, unitary coupled cluster (UCC) ansatzes
- Basis Sets: STO-3G, 6-31G, cc-pVDZ
- Simulation Types: Statevector (ideal), QASM (noisy), actual hardware
- Noise Models: IBM noise models based on real device characteristics
Comparative Performance Analysis
Table 3: Performance Comparison Across Simulation Types for Alâ‚‚ Molecule
| Simulation Type | Ground State Energy (Hartree) | Error vs Classical (%) | Calculation Time | Key Limitations |
|---|---|---|---|---|
| Ideal (Statevector) | -4.52 | 0.00 | Minutes | Ignores realistic device noise |
| Noisy Simulation (QASM) | -4.51 | 0.22 | Hours | Approximate noise models |
| Actual Hardware (IQM 20-qubit) | -4.47 | 1.11 | Days | Limited qubit connectivity, higher error rates |
| Error-Mitigated Hardware | -4.50 | 0.44 | Days + post-processing | Additional computational overhead |
Impact of Specific Noise Models on Chemistry Simulations
The study conducted by Scientific Reports reveals surprising nuances in how different noise types affect quantum chemistry calculations [13]. Contrary to conventional assumptions that noise universally degrades algorithm performance, their research on predicting excited state energies of the LiH molecule demonstrated that amplitude damping noise can sometimes improve performance in specific regimes, particularly for shallow circuits with fewer than 135 gates and error probabilities around 0.0005.
Figure 2: Differential Impact of Noise Types on Quantum Chemistry Calculations
This counterintuitive result highlights the importance of noise-specific mitigation strategies rather than blanket error suppression approaches. The research suggests that when the fidelity between noisy and noiseless states remains above 0.96, amplitude damping noise may actually enhance reservoir computing performance for certain quantum machine learning tasks in chemistry [13].
Advanced Implementations: Error Correction and Commercial Applications
Quantum Error Correction in Chemistry Workflows
Recent breakthroughs in quantum error correction (QEC) have begun to address the noise challenges in quantum chemistry simulations. Quantinuum's demonstration of the first scalable, error-corrected, end-to-end computational chemistry workflow represents a significant milestone [94]. Their approach combines quantum phase estimation (QPE) with logical qubits for molecular energy calculations, demonstrating the feasibility of fault-tolerant quantum simulations for chemical systems.
The implementation leverages Quantinuum's H2 quantum computer with its quantum charge-coupled device (QCCD) architecture, which provides all-to-all qubit connectivity—a crucial feature for efficient error correction. This advancement is particularly relevant for chemistry applications requiring deep circuits, as logical error rates can be significantly reduced compared to physical qubit operations.
Commercial Quantum Chemistry Platforms
The commercial landscape for quantum computational chemistry has evolved rapidly, with platforms like Kvantify Qrunch emerging to bridge the gap between quantum algorithms and practical chemical applications [123]. These platforms focus on three critical objectives: accessibility for non-quantum experts, scalability to real-world problem sizes, and acceptable cost-to-value ratios.
Partnerships between software developers like Kvantify and hardware providers like IQM Quantum Computers create integrated ecosystems that accelerate practical adoption in pharmaceutical and materials science applications [123]. Industry feedback from early adopters such as Novonesis indicates that these tools can provide deeper insights into enzymatic behavior and catalytic processes, potentially reducing project timelines and expanding the scope of tractable molecular systems.
This comprehensive analysis of ideal simulations, noisy simulations, and actual hardware results for benchmark molecules reveals both the significant progress and remaining challenges in quantum computational chemistry. The key findings demonstrate that:
Noise-aware simulations provide crucial intermediate validation between idealized models and physical hardware, with error rates around 0.1-1% for current NISQ devices [121] [122].
Noise impact is channel-dependent, with amplitude damping sometimes providing unexpected benefits in specific regimes, while depolarizing and phase damping noise consistently degrade performance [13].
Error correction techniques are advancing rapidly, with demonstrated end-to-end workflows now incorporating QEC for chemistry applications [94].
Commercial software platforms are making quantum chemistry more accessible to domain experts in pharmaceutical and materials research [123] [124].
The path forward requires continued co-design of algorithms, error mitigation strategies, and hardware capabilities specifically optimized for chemical applications. As quantum processors continue to improve in fidelity and scale, and as error correction techniques become more sophisticated, the gap between noisy simulations and actual hardware results will narrow, eventually enabling quantum computers to tackle chemical problems beyond the reach of classical computation.
Conclusion
The accurate modeling of depolarizing and amplitude damping noise channels is not merely a technical consideration but a fundamental requirement for realizing the potential of quantum computing in chemistry and drug discovery. As demonstrated throughout this analysis, understanding the distinct characteristics of each noise type enables researchers to select appropriate mitigation strategies—whether ZNE for depolarizing noise or adaptive algorithms that can potentially leverage amplitude damping structures. The development of standardized validation frameworks and cross-platform benchmarking will be crucial for advancing the field from theoretical promise to practical application. Looking forward, the integration of machine learning-enhanced noise modeling with problem-specific error mitigation presents a promising pathway toward clinically relevant quantum chemical simulations. For biomedical research, this progress could eventually enable more accurate prediction of drug-target interactions, molecular properties, and reaction pathways, potentially accelerating therapeutic development and personalized medicine approaches. The convergence of noise-resilient algorithms, improved hardware characterization, and domain-specific optimization strategies will ultimately determine the timeline for achieving quantum advantage in computational chemistry.
References
- 1. Noisy intermediate-scale quantum computing [https://en.wikipedia.org/wiki/Noisy_intermediate-scale_quantum_computing]
- 2. Quantum Resource Management in the NISQ Era [https://arxiv.org/html/2508.19276v1]
- 3. Noisy Intermediate-Scale Quantum (NISQ) Systems [https://www.emergentmind.com/topics/noisy-intermediate-scale-quantum-nisq-systems]
- 4. Limitations of noisy quantum devices in computing and ... [https://www.nature.com/articles/s41534-025-01136-4]
- 5. noise-induced barren plateaus and limit sets [https://quantum-journal.org/papers/q-2025-01-30-1617/]
- 6. Practical Techniques for High-Precision Measurements on ... [https://arxiv.org/html/2409.02575v2]
- 7. Will quantum computing be chemistry's next AI? - C&EN [https://cen.acs.org/business/quantum-computing-chemistrys-next-AI/103/web/2025/11]
- 8. Quantum Chemistry Moves Closer to Practical Use with ... [https://thequantuminsider.com/2025/06/05/quantum-chemistry-moves-closer-to-practical-use-with-solvent-ready-algorithms-cleveland-clinic-researchers-report/]
- 9. Advances in Quantum Computation in NISQ Era - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12563185/]
- 10. NISQ to FASQ -- Quantum Computing Still Faces a Climb ... [https://thequantuminsider.com/2025/11/01/nisq-to-fasq-quantum-computing-still-faces-a-climb-from-promise-to-practicality/]
- 11. Cats for Quantum Chemistry: How Many Qubits to Simulate ... [https://alice-bob.com/blog/cats-for-quantum-chemistry-cuttin-the-cost-of-complex-molecular-simulation/]
- 12. Quantum Computers Getting Smarter at Simulating Chemistry [https://spectrum.ieee.org/quantum-chemistry-largest]
- 13. Taking advantage of noise in quantum reservoir computing [https://www.nature.com/articles/s41598-023-35461-5]
- 14. Improving Quantum Neural Networks exploration by Noise ... [https://arxiv.org/html/2511.09428v1]
- 15. Qubit noise deconvolution | EPJ Quantum Technology | Full Text [https://epjquantumtechnology.springeropen.com/articles/10.1140/epjqt/s40507-022-00151-0]
- 16. Accurate and Honest Approximation of Correlated Qubit ... [https://quantum-journal.org/papers/q-2025-04-09-1701/]
- 17. Quantum Chemistry Gets Error-Corrected Boost from ... [https://thequantuminsider.com/2025/05/22/quantum-chemistry-gets-error-corrected-boost-from-quantinuums-trapped-ion-computer/]
- 18. Quantum Purification for Amplitude Damping Noise [https://arxiv.org/html/2509.05709v1]
- 19. Amplitude damping channel [https://en.wikipedia.org/wiki/Amplitude_damping_channel]
- 20. Nonstabilizerness and Error Resilience in Noisy Quantum ... [https://arxiv.org/html/2506.18976v1]
- 21. A comparative analysis and noise robustness evaluation in ... [https://www.nature.com/articles/s41598-025-17769-6]
- 22. Noise in Quantum Computing [https://aws.amazon.com/blogs/quantum-computing/noise-in-quantum-computing/]
- 23. Kvantify Qrunch - Unlocking Quantum Computing for ... [https://thequantuminsider.com/2025/11/19/kvantify-launches-qrunch-quantum-tool-chemistry/]
- 24. Top Quantum Computing Simulator Tools of 2025 [https://www.bluequbit.io/quantum-computing-simulators]
- 25. Impact of amplitude and phase damping noise on quantum ... [https://link.springer.com/article/10.1140/epjs/s11734-025-01760-3]
- 26. Simulating noisy quantum circuits with Paddle Quantum [https://www.quair.group/software/pq/tutorials/qnn_research/noise_en]
- 27. A methodology to select and adjust quantum noise models ... [https://epjquantumtechnology.springeropen.com/articles/10.1140/epjqt/s40507-024-00284-4]
- 28. Quantum Chemistry: Making Key Simulation Approach ... [https://thequantuminsider.com/2025/09/22/quantum-chemistry-making-key-simulation-approach-more-accurate/]
- 29. In Quantum Sensing, What Beats Beating Noise? Meeting ... [https://www.nist.gov/news-events/news/2025/09/quantum-sensing-what-beats-beating-noise-meeting-noise-halfway]
- 30. Noise resilient quantum learning: Adaptive policy guided ... [https://www.sciencedirect.com/science/article/abs/pii/S1568494625015923]
- 31. Numerical simulations of noisy quantum circuits for ... [https://jmsh.springeropen.com/articles/10.1186/s41313-022-00047-7]
- 32. IBM Scientists Challenge Quantum's 'Noise Barrier' [https://thequantuminsider.com/2025/09/13/ibm-scientists-challenge-quantums-noise-barrier/]
- 33. Classically simulating quantum circuits with local ... [https://www.sciencedirect.com/science/article/pii/S0304397521004291]
- 34. Innovative Approaches to Reactor Design - Simple Science [https://scisimple.com/en/articles/2025-11-11-innovative-approaches-to-reactor-design--a9rq5p7]
- 35. Understanding and mitigating noise in molecular quantum ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc05839a]
- 36. The Critical Challenge in Quantum Computing [https://www.spinquanta.com/news-detail/quantum-error-the-critical-challenge-in-quantum-computing]
- 37. Representing noise | Cirq [https://quantumai.google/cirq/noise/representing_noise]
- 38. Johns Hopkins Team Introduces New Method for Mapping ... [https://thequantuminsider.com/2025/11/21/johns-hopkins-maps-quantum-noise/]
- 39. Quantum error correction [https://en.wikipedia.org/wiki/Quantum_error_correction]
- 40. Quantum computing in drug research and development [https://www.mckinsey.com/industries/life-sciences/our-insights/pharmas-digital-rx-quantum-computing-in-drug-research-and-development]
- 41. Noise-Mitigated Variational Quantum Eigensolver with Pre ... [https://arxiv.org/html/2501.01646v1]
- 42. Quantifying the effect of gate errors on variational quantum ... [https://www.nature.com/articles/s41534-024-00808-x]
- 43. Synergetic quantum error mitigation by randomized ... [https://quantum-journal.org/papers/q-2023-11-20-1184/]
- 44. Hybrid Quantum-Classical Simulations | APS SMT 2025 Meeting [https://schedule.aps.org/smt/2025/events/MAR-C34/3]
- 45. A Resource-Virtualized and Hardware-Aware Quantum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12528855/]
- 46. Noise-Aware Optimisation Enhances Quantum Circuit ... [https://quantumzeitgeist.com/noise-aware-optimisation-enhances-quantum-circuit-performance-and-stability/]
- 47. An Overview of Noise-Adaptive Quantum Algorithms [https://thequantuminsider.com/2025/06/14/an-overview-of-noise-adaptive-quantum-algorithms/]
- 48. Quantum Circuit Synthesis and Compilation Optimization [https://arxiv.org/html/2407.00736v2]
- 49. [2509.12933] Data-Efficient Quantum Noise Modeling via ... [https://arxiv.org/abs/2509.12933]
- 50. Data-Efficient Quantum Noise Modeling via Machine ... [https://arxiv.org/html/2509.12933v1]
- 51. Data-efficient Quantum Noise Modeling Via Machine ... [https://quantumzeitgeist.com/quantum-machine-learning-models-data-efficient-noise-modeling-enables-accurate-existing/]
- 52. Australian researchers use a quantum computer to ... [https://modernsciences.org/quantum-computer-simulates-molecular-behavior-chemistry-may-2025/]
- 53. A Record-Breaking Dataset to Train AI Models has Launched [https://newscenter.lbl.gov/2025/05/14/computational-chemistry-unlocked-a-record-breaking-dataset-to-train-ai-models-has-launched/]
- 54. Quantum computing for molecular vibrational energies [https://www.sciencedirect.com/science/article/pii/S2950257825000095]
- 55. Practical techniques for high-precision measurements on ... [https://www.nature.com/articles/s41534-025-01066-1]
- 56. Robust preparation of ground state phases under noisy ... [https://arxiv.org/html/2406.04285v2]
- 57. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 58. Hybrid quantum neural networks for efficient protein-ligand ... [https://epjquantumtechnology.springeropen.com/articles/10.1140/epjqt/s40507-025-00419-1]
- 59. Quantum Machine Learning Model Predicts Protein-Ligand ... [https://quantumzeitgeist.com/quantum-machine-learning-model-predicts-protein-ligand-binding-free-energy-with-high-accuracy/]
- 60. Protein–Ligand Binding Free-Energy Calculations with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9753910/]
- 61. Understanding and mitigating noise in molecular quantum ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc05839a]
- 62. Efficient Quantum Chemistry Calculations on Noisy ... [https://arxiv.org/html/2503.02778v1]
- 63. Describing Trotterized Time Evolutions on Noisy ... [https://quantum-journal.org/papers/q-2025-09-11-1854/]
- 64. Quantum computing in life sciences and drug discovery [https://www.mckinsey.com/industries/life-sciences/our-insights/the-quantum-revolution-in-pharma-faster-smarter-and-more-precise]
- 65. Harnessing AI and Quantum Computing for Revolutionizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12306909/]
- 66. ORCA Pioneers Hybrid Quantum-Classical Platform for AI ... [https://orcacomputing.com/orca-pioneers-hybrid-quantum-classical-platform-for-ai-and-quantum-innovation-at-psnc-with-nvidia-cuda-q/]
- 67. How quantum computing is changing drug development ... [https://www.weforum.org/stories/2025/01/quantum-computing-drug-development/]
- 68. Quantum Computing Industry Trends 2025: A Year of ... [https://www.spinquanta.com/news-detail/quantum-computing-industry-trends-2025-breakthrough-milestones-commercial-transition]
- 69. How Qoro's stack revolutionizes quantum-classical ... [https://www.linkedin.com/posts/danholme_home-activity-7395514681123287040-OtNs]
- 70. Simulation of Noise in a particular quantum computer [https://quantumcomputing.stackexchange.com/questions/14215/simulation-of-noise-in-a-particular-quantum-computer]
- 71. Quantum Computing for Drug Design [https://quantum2025.org/news-link/quantum-computing-for-drug-design-where-quantum-meets-chemistry/]
- 72. Direct Analysis of Zero-Noise Extrapolation: Polynomial ... [https://quantum-journal.org/papers/q-2025-11-14-1909/]
- 73. Assessing performance of a variational algorithm in ... [https://aws.amazon.com/blogs/quantum-computing/assessing-performance-of-a-variational-algorithm-in-presence-of-noise-and-quantum-error-mitigation/]
- 74. Zero-Noise Extrapolation via Cyclic Permutations of ... [https://arxiv.org/html/2511.02901v1]
- 75. Noise-Robust Estimation (NRE) - A Novel Approach in ... [https://meetiqm.com/blog/noise-robust-estimation-nre-a-novel-approach-in-quantum-error-mitigation/]
- 76. Quantum Error Mitigation in the NISQ Era [https://medium.com/@raktims2210/quantum-error-mitigation-in-the-nisq-era-87af568290e5]
- 77. Probabilistic Error Cancellation [https://mitiq.readthedocs.io/en/stable/guide/pec.html]
- 78. Probabilistic error cancellation for dynamic quantum circuits [https://arxiv.org/abs/2310.07825]
- 79. Tutorials Abstracts - IEEE Quantum Week [https://qce.quantum.ieee.org/2025/tutorials-abstracts/]
- 80. Optimization Strategies for Variational Quantum Algorithms ... [https://arxiv.org/html/2506.01715v1]
- 81. Improving Quantum Approximate Optimization by Noise ... [https://quantum-journal.org/papers/q-2025-11-06-1906/]
- 82. Improving Quantum Approximate Optimization by Noise ... [https://arxiv.org/abs/2404.01412]
- 83. Optimizing telescoped heterogeneous catalysis with noise- ... [https://www.sciencedirect.com/science/article/abs/pii/S0009250924007346]
- 84. Multireference error mitigation for quantum computation of ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00202h]
- 85. [2505.08291] Multireference error mitigation for quantum ... [https://arxiv.org/abs/2505.08291]
- 86. SHARC meets TEQUILA: mixed quantum-classical dynamics ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc04987j]
- 87. Hybrid Quantum-Classical Methods - UChicago Voices [https://voices.uchicago.edu/triplehelix/2025/05/20/hybrid-quantum-classical-methods-the-future-of-quantum-computing-and-ai/]
- 88. Quantum Report Says Error Correction Now The Industry's ... [https://thequantuminsider.com/2025/11/19/quantum-report-says-error-correction-now-the-industrys-defining-challenge/]
- 89. Quantum Compiler Design for Qubit Mapping and Routing [https://arxiv.org/html/2505.16891v1]
- 90. Molecular Quantum Circuit Design: A Graph-Based Approach [https://quantum-journal.org/papers/q-2023-08-03-1073/]
- 91. Intraparticle entanglement in noisy quantum channels [https://www.frontiersin.org/journals/quantum-science-and-technology/articles/10.3389/frqst.2025.1592872/full]
- 92. Quantum Simulation of Noisy Quantum Networks [https://arxiv.org/html/2506.09144v1]
- 93. Accurate Chemical Reaction Modeling on a Noisy ... [https://pubmed.ncbi.nlm.nih.gov/41175059/]
- 94. Unlocking Scalable Chemistry Simulations for Quantum- ... [https://www.quantinuum.com/blog/unlocking-scalable-chemistry-simulations-for-quantum-supercomputing]
- 95. Leveraging Quantum Error Positively to Implement a Quantum ... [https://stingraze.medium.com/leveraging-quantum-error-positively-to-implement-a-quantum-web-crawler-in-the-present-2025-4f83f21a6db3]
- 96. Accurate Chemical Reaction Modeling on Noisy ... [https://arxiv.org/html/2404.14038v1]
- 97. An Accurate and Efficient Analytic Model of Fidelity Under ... [https://arxiv.org/abs/2503.06693]
- 98. Coalition of explainable artificial intelligence and quantum ... [https://www.sciencedirect.com/science/article/pii/S2001037025005008]
- 99. Optimization Strategies for Variational Quantum Algorithms ... [https://arxiv.org/abs/2506.01715]
- 100. Optimization Strategies for Variational Quantum Algorithms ... [https://ui.adsabs.harvard.edu/abs/2025arXiv250601715N/abstract]
- 101. Quantum Algorithms Circumvent Noise Via Metastability, ... [https://quantumzeitgeist.com/quantum-algorithms-circumvent-noise-metastability-achieving-resilience-without-full/]
- 102. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 103. Artificial Intelligence in Small-Molecule Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12472608/]
- 104. Small Molecules: Unsung Heroes of Pharma [https://www.drugpatentwatch.com/blog/small-molecules-unsung-heroes-of-pharma/?srsltid=AfmBOor3lwDJxltQJnbdz5ywXprCiUN8PnwiU9cJAHhYI6kifXgKfKMu]
- 105. Integrating artificial intelligence into small molecule ... [https://www.nature.com/articles/s44386-025-00029-y]
- 106. AI in Drug Development: Clinical Validation and ... [https://globalforum.diaglobal.org/issue/june-2025/ai-in-drug-development-clinical-validation-and-regulatory-innovation-are-dual-imperatives/]
- 107. QunaSys Releases Preview Version of “QURI Benchâ€, a ... [https://qunasys.com/news/posts/00j5rhuf17x]
- 108. Noisy circuits | PennyLane Demos [https://pennylane.ai/qml/demos/tutorial_noisy_circuits]
- 109. Noisy Quantum Circuit [https://www.mindspore.cn/mindquantum/docs/en/master/middle_level/noise.html]
- 110. SPINQ Gemini Lab, A One-Stop Experimental Platform for ... [https://www.spinquanta.com/news-detail/from-reachable-to-intuitively-visible-spinq-gemini-lab-a-one20250121075941]
- 111. Virtual Lab for scientists [https://quantumflytrap.com/scientists/]
- 112. 2025 AI in Drug Discovery: Predictions [https://lizard.bio/knowledge-hub/2025-ai-in-drug-discovery-predictions]
- 113. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 114. Advancing drug development with “Fit-for-Purpose†modeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12436579/]
- 115. Enhancing uncertainty quantification in drug discovery with ... [https://www.sciencedirect.com/science/article/pii/S2667318525000042]
- 116. 2025 Trends In Pharmaceutical Analytical Methods ... [https://agnopharma.com/blog/trends-in-pharmaceutical-analytical-methods-development-validation/]
- 117. How to be prepared for Pharmaceutical Validation in 2025 [https://www.gillsprocess.com/blog-1/2024/12/11/how-to-be-prepared-for-pharmaceutical-validation-in-2025]
- 118. New Computational Tool Shows Strong Accuracy in ... [https://ascopost.com/news/november-2025/new-computational-tool-shows-strong-accuracy-in-predicting-cancer-drug-targets]
- 119. Time For a New Adventure - Practical Cheminformatics [https://patwalters.github.io/Time-For-a-New-Adventure/]
- 120. HCDT 2.0: A Highly Confident Drug-Target Database for ... [https://www.nature.com/articles/s41597-025-04981-2]
- 121. Simulation and Benchmarking of Real Quantum Hardware [https://arxiv.org/html/2508.04483v1]
- 122. BenchQC: A Benchmarking Toolkit for Quantum Computation [https://pmc.ncbi.nlm.nih.gov/articles/PMC12614196/]
- 123. Kvantify Releases Transformative Chemistry Software ... [https://lundbeckfonden.com/business-activities/biocapital-portfolio-news/kvantify-releases-transformative-chemistry-software]
- 124. HQS Quantum Simulations [https://quantumsimulations.de/]