Benchmarking Quantum Chemistry: A Practical Guide to Validating Conformational Energies with DFT and Post-HF Methods in Drug Discovery
Accurate calculation of conformational energies is a cornerstone of reliable computational chemistry in drug design, impacting everything from docking poses to property prediction.
Benchmarking Quantum Chemistry: A Practical Guide to Validating Conformational Energies with DFT and Post-HF Methods in Drug Discovery
Abstract
Accurate calculation of conformational energies is a cornerstone of reliable computational chemistry in drug design, impacting everything from docking poses to property prediction. This article provides a comprehensive guide for researchers and development professionals on validating these critical energies. We first explore the fundamental importance of conformational analysis and establish DLPNO-CCSD(T) as the modern reference 'gold standard.' The guide then details best-practice protocols for applying DFT and post-Hartree-Fock methods, highlighting common pitfalls and optimization strategies for robust workflows. Finally, we present a systematic framework for the comparative validation of different computational methods against high-level benchmarks, empowering scientists to select and trust the right tool for their specific drug discovery project.
The Critical Role of Conformational Energies in Drug Design: From Fundamentals to Gold Standards
Why Conformational Energy Accuracy Matters for Bioactive Pose Prediction and Property Calculation
In rational drug design, the biological activity and physicochemical properties of a molecule are not determined by a single static structure, but by an ensemble of accessible three-dimensional conformations. The accurate prediction of this conformational landscape and its associated energetics is a cornerstone of computational chemistry, directly impacting the success of structure-based and ligand-based drug discovery campaigns. The energy differences between conformers are often subtle, yet they govern the population of specific states, including the bioactive conformation that a molecule adopts when bound to its protein target. Inaccuracies in calculating these conformational energies can lead to failures in predicting binding poses, estimating binding affinities, and optimizing lead compounds. This guide examines the critical importance of conformational energy accuracy, comparing the performance of various computational methods and providing a framework for their practical application in drug discovery.
Performance Comparison of Conformational Sampling Methods
Force Fields and Solvation Models in Bioactive Conformation Retrieval
The ability of a computational method to generate a conformational ensemble that includes a structure closely resembling the experimentally observed bioactive pose is a fundamental benchmark. A study evaluating drug-like ligands from protein-ligand complexes in the Protein Data Bank (PDB) investigated the performance of various force fields and solvation settings for this task.
Table 1: Impact of Force Field and Solvation on Bioactive Conformation Retrieval (Root Mean Square Deviation < 1.0 Å)
| Method Category | Specific Method | Performance in Bioactive Conformation Retrieval | Key Findings |
|---|---|---|---|
| Force Fields | Various (e.g., OPLS_2005) | Only small differences in likelihood | Modern force fields show comparable performance in sampling ability. [1] |
| Solvation Models | Implicit Water (GB/SA) | High likelihood | Crucial for achieving low RMSD (<1.0 Å) to crystal pose. [1] |
| Solvation Models | Vacuum / Other Solvents | Lower likelihood | Less effective for achieving high geometric accuracy. [1] |
| Search Parameters | Duplicate Removal (RMSD ~0.6 Å) | Large impact | Stringent duplicate removal is critical for success. [2] |
| Search Parameters | High Energy Cut-off (e.g., 5 kcal/mol) | High impact | A generous energy window increases retrieval rate. [2] |
The findings indicate that while the choice of modern force field introduces only minor variations, the inclusion of an appropriate solvation model, particularly implicit water, is critically important. Furthermore, the parameters controlling the conformational search itself, such as the thresholds for considering two conformers as duplicates and the energy window for saving conformers, have a substantial impact on the success rate. [1] [2]
Quantum Mechanical Methods for High-Accuracy Conformational Energies
For higher accuracy, especially when dealing with non-covalent interactions or electronic properties, quantum mechanical (QM) methods are essential. However, a direct trade-off exists between computational cost and accuracy.
Table 2: Performance and Cost of Quantum Mechanical Workflows for Conformational Energies
| Method / Workflow | Typical Application | Relative Cost | Key Advantages & Limitations |
|---|---|---|---|
| Semi-Empirical (GFN2-xTB) | Initial conformational sampling | Low | Fast; good for initial ensemble generation, but energy rankings may differ from DFT. [3] [4] |
| Composite "3c" Methods (B97-3c) | Ensemble re-ranking and pre-optimization | Medium | Good balance of speed and accuracy; reduces spurious minima from GFN2-xTB. [4] |
| Density Functional Theory (ωB97X-D) | High-quality final energies & geometries | High | High accuracy for energies and geometries; considered a benchmark for many applications. [4] |
| Workflow (CREST → B97-3c → DFT) | Full DFT-quality ensemble generation | High | Efficiently produces reliable ensembles; avoids coalescence of spurious conformers. [4] |
The performance of DFT functionals themselves can be benchmarked for specific properties. For instance, assessing core-electron binding energies (CEBEs), which are sensitive to the electronic environment, reveals functional-dependent accuracy.
Table 3: DFT Functional Performance for Core-Electron Binding Energy (CEBE) Prediction
| DFT Functional | Performance (RMSD for C1s CEBEs) | Key Insight |
|---|---|---|
| PW86x-PW91c | 0.1735 eV | Good general performance. [5] |
| mPW1PW | ~0.132 eV (AAD) | Improved accuracy for polar C-X bonds (X=O, F). [5] |
| PBE50 | ~0.132 eV (AAD) | Improved accuracy for polar C-X bonds; highlights role of HF exchange. [5] |
Experimental Protocols for Conformational Benchmarking
Protocol 1: Evaluating Bioactive Conformation Retrieval
This protocol is adapted from studies that benchmark conformational search tools against high-resolution crystal structures of protein-ligand complexes. [1] [2]
- Ligand Curation: Select a diverse set of drug-like ligands from the PDB. Key criteria include:
- High-resolution complexes (e.g., ≤ 2.0 Å).
- No covalent bonding to the protein.
- Drug-like properties (MW 150-650, ≤ 15 rotatable bonds).
- Exclusion of common cofactors and crystallization agents. [1]
- Ligand Preparation: Add hydrogen atoms using a curated library, predict the most probable protonation state at the crystallization pH, and assign formal charges. [1]
- Conformational Search: Perform a two-step conformational search for each ligand.
- Step 1 (Debiasing): Use a force field like OPLS_2005 with a GB/SA water model and a mixed MCMM/Low-Mode algorithm. Generate a large pool of conformers (e.g., up to 1000) with a high energy cut-off (e.g., 50.0 kcal/mol from the global minimum). [1]
- Step 2 (Production): Use the generated conformers as input for the method being tested (e.g., OMEGA, CREST, MacroModel), varying key parameters like the RMSD duplicate cut-off and maximum number of output conformers. [2]
- Analysis: For each ligand's conformer pool, calculate the heavy-atom Root Mean Square Deviation (RMSD) of every generated conformer to the experimental bioactive conformation. The performance metric is the best achievable RMSD and the success rate in retrieving a conformation below a defined threshold (e.g., 1.0 Å or 0.5 Å). [1] [2]
Protocol 2: Generating DFT-Quality Conformational Ensembles
This modern workflow leverages efficient semi-empirical sampling followed by higher-level theory refinement to produce accurate conformational ensembles for property prediction. [4]
- Initial Ensemble Generation: Use the CREST program with the GFN2-xTB method to generate a broad, initial set of conformers. CREST uses RMSD-biased metadynamics to thoroughly explore the conformational space, typically returning all conformers within a ~6 kcal/mol energy window. [3] [4]
- Intermediate Re-optimization and Re-ranking: Re-optimize the entire CREST-generated ensemble using a cost-effective composite DFT method (e.g., B97-3c). This step significantly improves geometries and energy rankings over the GFN2-xTB level while remaining computationally feasible for large ensembles. [4]
- Duplicate Removal: Identify and discard duplicate conformers from the re-optimized set based on an RMSD threshold.
- High-Level DFT Refinement: Re-optimize the remaining unique, low-energy conformers using a higher-level DFT method (e.g., ωB97X-D/def2-SVP). This provides final, high-quality geometries. [4]
- Final Energetics Calculation: Perform a final single-point energy calculation on the refined geometries using an even larger basis set (e.g., ωB97X-V/def2-QZVPP) to obtain highly accurate relative energies. Vibrational frequency calculations can be used to derive thermodynamic corrections and free energies. [4]
Diagram 1: Workflow for generating DFT-quality conformational ensembles, balancing thorough sampling with high accuracy.
The Scientist's Toolkit: Essential Research Reagents and Software
Table 4: Key Software and Computational Tools for Conformational Analysis
| Tool Name | Category | Primary Function | Relevance to Conformational Energy |
|---|---|---|---|
| MacroModel | Software Suite | Conformational Search & Minimization | Implements various force fields and algorithms (e.g., MCMM) for generating conformers in solvation. [1] |
| OMEGA | Software | Rule-based Conformer Generation | Rapidly generates large conformational ensembles; performance depends on parameters like duplicate RMSD. [2] |
| CREST | Software | Conformer-Rotamer Ensemble Sampling | Uses GFN2-xTB and metadynamics for extensive sampling, forming the basis for high-level workflows. [3] [4] |
| GEOM Dataset | Database | Energy-Annotated Molecular Conformations | Provides a large benchmark of conformers with semi-empirical and DFT energies for method development. [3] |
| FlexiSol | Database | Solvation Benchmark with Conformer Ensembles | Enables testing of solvation models on flexible molecules using conformational ensembles. [6] |
| ORCA | Software | Quantum Chemistry | Performs high-level DFT and composite method calculations for final geometry and energy refinement. [4] |
Accurate conformational energies are not merely an academic exercise but a practical necessity in modern computational drug discovery. The choice of computational method, from classical force fields to advanced DFT workflows, directly influences the reliability of bioactive pose prediction and the calculation of conformationally dependent molecular properties. As benchmarked against experimental data, method performance varies significantly, with key factors being the treatment of solvation, the parameters of conformational search, and the underlying level of theory used for energy evaluation. The integration of robust experimental protocols, modern software tools, and curated benchmark datasets, as detailed in this guide, provides a pathway for researchers to critically evaluate and apply these methods. This ensures that the critical role of molecular conformation is accurately captured, thereby de-risking the drug design process and increasing the likelihood of success in bringing new therapeutics to patients.
In the fields of computational chemistry and computer-aided drug design, accurately predicting the energy and three-dimensional structure of molecules is fundamental. The "gold standard" for such calculations has long been the coupled-cluster with single, double, and perturbative triple excitations (CCSD(T)) method, celebrated for its excellent agreement with experimental data [7]. However, its overwhelming computational cost, which scales to the seventh power with system size, renders it prohibitive for the large, drug-like molecules of practical interest [7] [8].
The breakthrough came with the development of local correlation approximations. Among these, the Domain-based Local Pair Natural Orbital (DLPNO) approach has made it feasible to perform CCSD(T) calculations on molecules containing hundreds of atoms [9] [8]. The DLPNO-CCSD(T) method achieves this by restricting electron correlation to local domains and using compressed sets of virtual orbitals for each electron pair, dramatically reducing computational cost while striving to retain the accuracy of its canonical predecessor [7] [10]. This article explores how DLPNO-CCSD(T) has established itself as a modern benchmark for calculating conformational energies and non-covalent interactions, providing a reliable standard for validating force fields, density functional theory (DFT), and other quantum mechanical methods in drug discovery research [9].
Performance Benchmarks: How DLPNO-CCSD(T) Compares
Extensive benchmarking studies have systematically evaluated the performance of DLPNO-CCSD(T) against canonical CCSD(T) and other computational methods across various chemical systems.
Accuracy Against Canonical CCSD(T)
The primary validation for any approximate method is its agreement with the established gold standard. Research consistently shows that with appropriate settings, DLPNO-CCSD(T) delivers exceptional accuracy:
- For hydrogen atom transfer (HAT) reactions in closed-shell systems, the standard deviation in calculated barrier heights between DLPNO-CCSD(T) and canonical CCSD(T) was found to be less than 0.2 kcal mol⁻¹ across multiple basis sets [7].
- For water clusters (H₂O)ₙ=₃–₇, DLPNO-CCSD(T) relative energies compared to explicitly correlated CCSD(T)-F12b benchmarks showed mean absolute differences (MADs) of ≤ 0.13 kcal/mol when triple-zeta and larger basis sets were employed [11].
- For atmospheric molecular clusters, DLPNO-CCSD(T0)-F12 with a tightPNO criterion demonstrated remarkable precision, with a mean deviation of 0.10 kcal/mol and a maximum deviation of 0.20 kcal/mol from the CCSD(F12*)(T)/CBS reference [10].
Performance as a Reference for Conformational Energies
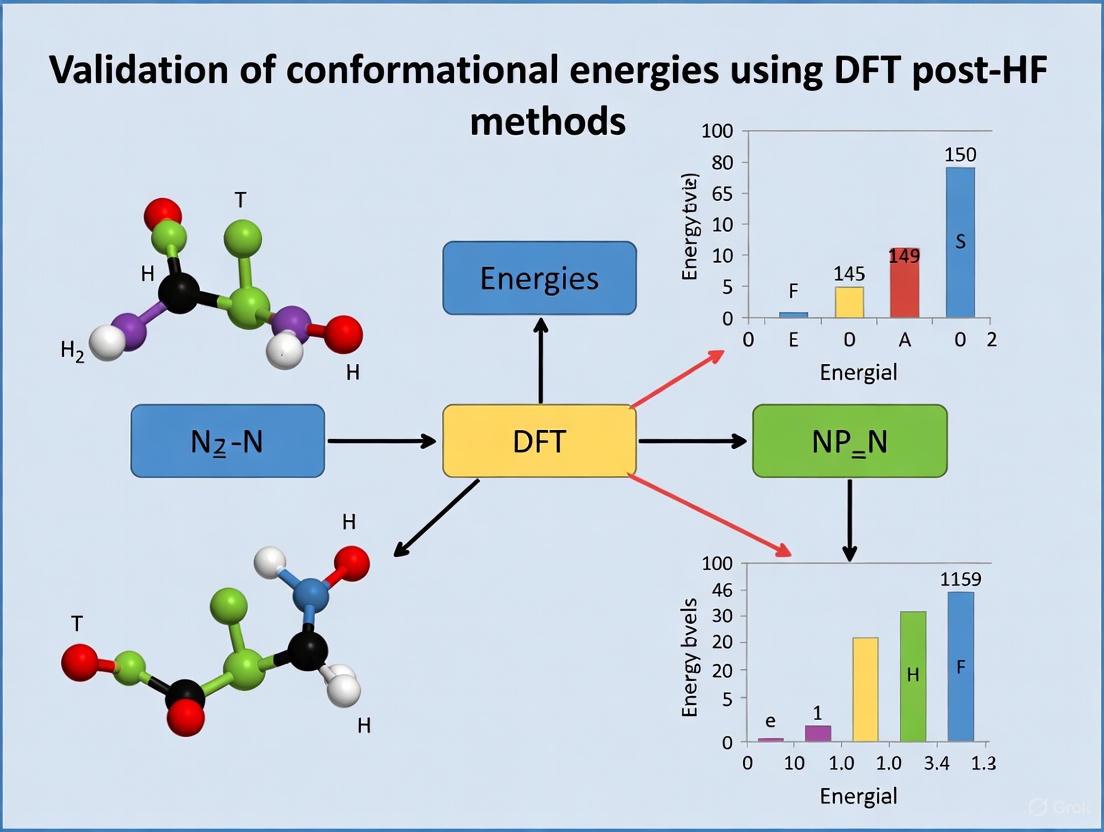
A comprehensive 2023 study evaluated the accuracy of various force fields and quantum mechanics methods for calculating the conformational energies of 145 reference organic molecules, using DLPNO-CCSD(T) as the reference [9]. The results underscore its role as a definitive benchmark. The table below summarizes the performance of different method classes against the DLPNO-CCSD(T) reference.
Table 1: Mean Error in Conformational Energies of Organic Molecules vs. DLPNO-CCSD(T) Reference [9]
| Method Class | Specific Method | Mean Error (kcal mol⁻¹) |
|---|---|---|
| Wavefunction Methods | MP2 | 0.35 |
| B3LYP | 0.69 | |
| HF | 0.81 – 1.00 | |
| Advanced Force Fields | MMFF94 | 1.30 |
| MM3-00 | 1.28 | |
| MM3-96 | 1.40 | |
| Traditional Force Fields | MMX | 1.77 |
| MM+ | 2.01 | |
| MM4 | 2.05 | |
| Generic Force Fields | DREIDING | 3.63 |
| UFF | 3.77 |
This study highlights that while certain ab initio methods like MP2 show excellent agreement, commonly used force fields can introduce significant errors. The DLPNO-CCSD(T) method provides the high-accuracy reference data needed to identify these discrepancies and guide the parameterization of improved force fields [9].
Experimental Protocols: Implementing DLPNO-CCSD(T) Calculations
To achieve high accuracy with DLPNO-CCSD(T), researchers must follow careful computational protocols. The workflow below outlines the key stages of a typical study using DLPNO-CCSD(T) to benchmark conformational energies.
Diagram 1: DLPNO-CCSD(T) Benchmarking Workflow
Detailed Methodology
System Preparation and Geometry Optimization: The process begins with generating accurate molecular structures.
- Initial Conformer Generation: For drug-like molecules, diverse low-energy conformers are typically generated using tools like OMEGA or RDKit, often employing force fields like MMFF94 for initial screening [9].
- Geometry Optimization: These initial structures are then optimized at a higher level of theory to locate true energy minima. Proven methods include:
Reference Energy Calculation with DLPNO-CCSD(T): Single-point energy calculations at the optimized geometries provide the benchmark energies.
- Method and Keywords: The DLPNO-CCSD(T) method is used, often with the
TightPNOsetting to enhance accuracy for sensitive properties like conformational energies [10] [12]. For open-shell systems or those with potential multireference character, adjusting theTcutPNOparameter may be necessary [7]. - Basis Sets: Large basis sets from Dunning's correlation-consistent series (e.g.,
cc-pVTZ,cc-pVQZ) are standard. Diffuse functions are frequently added to heavy atoms (e.g.,aug-cc-pVTZ) to better capture non-covalent interactions [11]. - Explicit Correlation (F12): Using the explicitly correlated F12 variant (e.g., DLPNO-CCSD(T)-F12) significantly accelerates basis set convergence, allowing the use of smaller basis sets to achieve near-complete-basis-set (CBS) limit accuracy [10].
- Extrapolation to CBS Limit: For the highest accuracy, energies are computed with a series of basis sets of increasing size (e.g., triple-, quad-, and pentuple-zeta). These results are then extrapolated to the CBS limit using established functions to eliminate residual basis set error [11].
- Method and Keywords: The DLPNO-CCSD(T) method is used, often with the
Benchmarking Other Methods: The DLPNO-CCSD(T)/CBS energies serve as the reference to evaluate the performance of other methods. For each conformer, the energy difference relative to the global minimum is calculated using both the reference method and the method being tested. Statistical measures like Mean Absolute Deviation (MAD) and Root-Mean-Square Deviation (RMSD) are then computed to quantify performance [9].
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table details key computational "reagents" and resources essential for conducting research with DLPNO-CCSD(T).
Table 2: Essential Research Reagents and Computational Tools
| Item/Software | Function in Research | Key Considerations |
|---|---|---|
| ORCA | A widely used software package for ab initio calculations; features a highly efficient, widely cited implementation of the DLPNO-CCSD(T) method. | The main platform for DLPNO calculations; developed by the Neese group [7]. |
| Molpro | Another quantum chemistry software that offers local coupled-cluster methods like PNO-LCCSD(T)-F12, used for high-accuracy reference calculations. | Often used for canonical CCSD(T) and explicitly correlated methods [8]. |
Basis Sets (e.g., cc-pVNZ, aug-cc-pVNZ) |
Mathematical sets of functions used to represent molecular orbitals; larger sets (higher N) increase accuracy but also computational cost. | Augmented sets (aug-) are critical for non-covalent interactions [11]. The haNZ convention (e.g., haTZ) uses different basis sets for H and heavy atoms [11]. |
PNO Settings (TightPNO, NormalPNO) |
Computational parameters that control the size of the Pair Natural Orbitals, trading between accuracy and speed. | TightPNO or vTightPNO settings are recommended for benchmarking to minimize errors in energy differences [10] [12]. |
DFT Functionals (e.g., ωB97X-D) |
Used for efficient geometry optimization of molecular structures prior to high-level DLPNO-CCSD(T) single-point energy calculations. | Functionals with dispersion corrections are crucial for obtaining correct geometries of organic molecules and non-covalent clusters [11]. |
Force Fields (e.g., MMFF94, MM3) |
Classical potentials used for initial conformational sampling and rapid energy estimation; their parameters are often refined against DLPNO-CCSD(T) data. | Performance varies significantly; DLPNO-CCSD(T) benchmarks identify which force fields are suitable for a given class of molecules [9]. |
DLPNO-CCSD(T) has unequivocally emerged as a modern gold standard in computational chemistry, particularly for generating benchmark-quality data on conformational energies and non-covalent interactions relevant to drug discovery. By providing a near-indistinguishable alternative to canonical CCSD(T) at a fraction of the cost, it enables the rigorous validation and improvement of more approximate methods like density functional theory and molecular mechanics force fields [9] [11]. As computational efforts increasingly focus on large, pharmaceutically relevant molecules, DLPNO-CCSD(T) stands as the critical benchmark, ensuring that the predictive models used in computer-aided drug design are built upon a foundation of chemical accuracy.
Fragment-based drug discovery (FBDD) has established itself as a powerful and complementary approach to high-throughput screening (HTS) for identifying novel drug leads [13]. This methodology involves screening low molecular weight compounds (typically ≤ 20 heavy atoms) against biological targets, followed by structure-guided optimization of these weakly binding hits into potent drug candidates [14]. The fundamental advantage of FBDD lies in its efficient sampling of chemical space; a small library of 1,000-2,000 fragments can provide proportionately greater coverage than much larger HTS libraries comprising larger, more complex molecules [13]. This approach has already yielded significant clinical successes, including six marketed drugs such as vemurafenib, venetoclax, and sotorasib—the latter highlighting FBDD's utility against targets like KRASG12C that were long considered "undruggable" [13] [14].
The selection of appropriate organic fragments is paramount for constructing reliable validation sets to benchmark computational methods in drug discovery. These fragments serve as critical test cases for evaluating the performance of quantum chemical methods, including Density Functional Theory (DFT) and post-Hartree-Fock (post-HF) approaches, in predicting conformational energies, binding affinities, and other physiochemical properties essential for drug development [15] [16]. A well-designed fragment validation set must encompass diverse chemical space while maintaining pharmaceutical relevance, enabling robust assessment of computational protocols against experimental data.
Defining Key Organic Fragments and Their Properties
Characteristics of Optimal Fragments
In FBDD, fragments are defined as small organic molecules with simple chemical structures. While historically guided by the "Rule of Three" (molecular weight ≤ 300 Da, H-bond donors ≤ 3, H-bond acceptors ≤ 3, cLogP ≤ 3), modern fragment libraries often include compounds that strategically violate these criteria to access novel chemical space [13]. Successful fragments typically exhibit high ligand efficiency (binding energy per heavy atom), enabling efficient optimization during later stages of drug development. Despite their small size, fragments form high-quality, "atom-efficient" interactions with protein targets, making them ideal starting points for medicinal chemistry campaigns [13].
The concept of fragment sociability has emerged as a crucial consideration in fragment selection. Sociable fragments contain well-established synthetic growth vectors and have numerous commercially available analogues, facilitating rapid structure-activity relationship (SAR) exploration during optimization [17]. Conversely, unsociable fragments lack robust synthetic methodology or accessible analogues, often impeding hit-to-lead progression despite promising initial binding [17].
Advantages Over Traditional Screening Approaches
Compared to conventional HTS, FBDD offers several distinct advantages beyond efficient chemical space sampling. Fragment hits typically display lower molecular complexity, reducing the probability of suboptimal interactions or steric clashes with target proteins [13]. Additionally, fragment screening provides valuable insights into target "druggability" and can identify binding hotspots at protein-protein interfaces or allosteric sites that are often challenging to address with larger compounds [13]. The weak affinities of initial fragment hits (typically in the μM-mM range) necessitate highly sensitive detection methods, including surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), and X-ray crystallography, with orthogonal techniques frequently employed for hit validation [13] [18].
Methodologies for Building Representative Validation Sets
Experimental Protocols for Fragment Screening and Validation
Building a comprehensive fragment validation set requires integration of multiple experimental techniques to thoroughly characterize fragment-target interactions. The following workflow outlines a robust protocol for experimental fragment validation:
Experimental Workflow for Fragment Validation Set Construction
Primary Screening Using Biophysical Methods
Initial fragment screening employs sensitive biophysical techniques capable of detecting weak interactions (Kd values in the μM-mM range). Surface plasmon resonance (SPR) provides real-time kinetic data on fragment binding, including association and dissociation rates [18]. Protein-observed NMR, particularly 19F NMR, offers a rapid method for fragment screening and can detect binding even for challenging targets like GPCRs [18]. These primary screens typically yield hit rates of 0.5-5%, significantly higher than HTS campaigns [13].
Hit Validation with Orthogonal Techniques
Confirmed hits from primary screening undergo validation using orthogonal biophysical methods to eliminate false positives and pan-assay interference compounds (PAINS). Differential scanning fluorimetry (DSF) and microscale thermophoresis (MST) provide additional confirmation of binding, each with distinct sensitivity profiles and potential biases [18]. This multi-technique approach ensures robust hit identification; studies reveal that different detection methods often yield surprisingly low overlap in hit lists, highlighting the importance of orthogonal validation [18].
Structural Characterization via X-ray Crystallography
High-resolution X-ray crystallography represents the gold standard in FBDD, providing atomic-level details of fragment binding modes [17]. This structural information is crucial for identifying optimal growth vectors—specific positions on the fragment scaffold where chemical modifications can enhance potency and properties [17]. For example, in a urokinase-type plasminogen activator (uPA) program, X-ray structures guided the selection of fragments with accessible growth vectors toward the catalytic triad, enabling significant affinity improvements [17].
Structure-Activity Relationship (SAR) Expansion
Sociable fragments with commercially available analogues enable rapid SAR exploration through purchase and testing of related compounds [17]. For instance, the fragment hit mexiletine in the uPA program had over 100 commercially available analogues, facilitating comprehensive mapping of the binding pocket and resulting in a potent lead compound (IC50 = 0.07 μM) [17]. This empirical SAR data provides invaluable validation for computational predictions of binding energies and molecular properties.
Computational Approaches for Fragment Validation
Computational methods play an increasingly important role in fragment validation, complementing experimental techniques. The integration of machine learning (ML) and quantum mechanical calculations enables prediction of fragment properties, binding modes, and synthetic accessibility.
Table 1: Computational Methods for Fragment Validation
| Method Category | Specific Approaches | Application in Fragment Validation | Key Considerations |
|---|---|---|---|
| Quantum Chemical Methods | DFT (ωB97M-D3(BJ), B3LYP-D3, ωB97X-D), MP2 | Conformational energy ranking, tautomer stability, protonation state prediction | Functional/basis set selection crucial; dispersion corrections essential for accuracy [16] [19] |
| Machine Learning Potentials | Neural network potentials, ANI models, QDπ dataset | Rapid energy evaluation, conformational sampling, property prediction | Training data quality determines transferability; active learning reduces redundancy [15] |
| Descriptor Analysis | DompeKeys (DK), Molecular Anatomy, ECFP | Chemical space mapping, functional group identification, toxicity alert detection | Hierarchical descriptors capture different complexity levels [20] |
| Conformer Generation | CREST, GEOM dataset, RDKit stochastic methods | Ensemble property prediction, Boltzmann-weighted averages | Coverage of thermally accessible conformers critical for accuracy [3] |
The QDπ dataset represents a significant advance for computational validation, incorporating 1.6 million molecular structures with energies and forces calculated at the ωB97M-D3(BJ)/def2-TZVPPD level of theory [15]. This dataset employs active learning strategies to maximize chemical diversity while minimizing redundant information, making it particularly valuable for developing universal machine learning potentials applicable to drug-like molecules [15].
The GEOM dataset provides another essential resource, containing 37 million molecular conformations for over 450,000 molecules, with specific annotation for 1,511 species with BACE-1 inhibition data [3]. This dataset enables benchmarking of models that predict properties from conformer ensembles, crucial for accurate representation of flexible fragments under physiological conditions.
Commercial Fragment Libraries and Their Characteristics
Several commercial fragment libraries are available, each with distinct properties and coverage of chemical space. While these provide excellent starting points, they often require curation and filtering to optimize diversity and pharmaceutical relevance.
Table 2: Comparison of Fragment Libraries and Validation Resources
| Library/Resource | Size (Compounds) | Key Characteristics | Applications in Validation | Notable Features |
|---|---|---|---|---|
| Commercial Fragment Libraries | 1,000-5,000 | MW ≤ 300, Ro3 compliance, varying sp3 character | Primary screening, hit identification | Often include "3D-shaped" fragments with enhanced sp3 character [13] |
| QDπ Dataset [15] | 1.6 million structures | ωB97M-D3(BJ)/def2-TZVPPD level, 13 elements | ML potential training, conformational energy validation | Active learning strategy minimizes redundancy while maximizing diversity |
| GEOM Dataset [3] | 37 million conformers (450k molecules) | CREST/GFN2-xTB sampling, experimental bioactivity data | Conformer ensemble prediction, Boltzmann-weighted properties | Includes explicit solvation data for BACE inhibitors |
| DompeKeys (DK) [20] | 1,064 curated SMARTS | Hierarchical substructure definition (5 complexity levels) | Chemical space mapping, functional group analysis | Specifically designed for pharmaceutical applications |
Best Practices for DFT Protocol Selection in Fragment Validation
Accurate computational validation of fragments requires careful selection of quantum chemical methods. Benchmark studies demonstrate that method choice significantly impacts prediction accuracy for conformational energies and other key properties.
Computational Protocol for Fragment Conformational Analysis
Evaluation of quantum chemical methods using A-value (conformational energy difference in substituted cyclohexanes) estimation provides valuable benchmarking data [19]. Studies reveal that B3LYP without dispersion correction significantly overestimates A-values, highlighting the importance of proper treatment of London dispersion forces [19]. Functionals like ωB97X-D and M06-2X generally provide better performance, though systematic errors may occur for specific substituents like tert-butyl groups [19]. Solvation effects, incorporated via implicit models like PCM, prove particularly important for polar substituents, reducing A-values by approximately 0.15 kcal mol-1 on average [19].
Table 3: Essential Research Reagent Solutions for Fragment-Based Discovery
| Resource Category | Specific Tools/Solutions | Function in Fragment Research | Key Providers/Platforms |
|---|---|---|---|
| Biophysical Screening | SPR (Biacore platforms), NMR spectrometers, MST | Detection of weak fragment binding, kinetic parameter determination | GE Healthcare, Bruker, NanoTemper |
| Structural Biology | X-ray crystallography platforms, Cryo-EM | High-resolution structure determination of fragment-protein complexes | Rigaku, Thermo Fisher Scientific |
| Chemical Databases | ZINC, eMolecules, CAS | Source of commercially available fragments and analogues | Multiple commercial suppliers |
| Computational Chemistry | Gaussian, ORCA, CREST, RDKit | Quantum chemical calculations, conformer generation, property prediction | Academic and commercial software |
| Specialized Libraries | Covalent fragment libraries, 3D-shaped fragments, Natural product-derived fragments | Access to underrepresented chemical space for challenging targets | Enamine, Life Chemicals, WuXi AppTec |
The construction of representative validation sets for organic fragments in drug-like molecules requires integrated experimental and computational approaches. Key considerations include fragment sociability for efficient optimization, comprehensive conformational sampling to account for molecular flexibility, and robust quantum chemical methods with proper treatment of non-covalent interactions. Publicly available resources like the QDπ and GEOM datasets provide essential benchmarking data for method development and validation.
Emerging trends in fragment-based discovery include increased incorporation of covalent fragments, expanded use of machine learning for hit identification and optimization, and growing emphasis on 3D-shaped fragments with enhanced sp3 character for challenging targets [13] [14]. As these methodologies continue to evolve, well-validated fragment sets will remain indispensable for advancing computational drug discovery and tackling previously "undruggable" targets.
Molecular mechanics force fields provide the foundational framework for simulating the structures, motions, and interactions of biological macromolecules and drug-like compounds in full atomic detail. The accuracy of these simulations, particularly in computer-aided drug design (CADD), is critically dependent on the force field chosen. Force fields are computational models that use mathematical functions and parameter sets to calculate the potential energy of a system based on atomic coordinates. Their functional form typically includes terms for bonded interactions (bonds, angles, dihedrals) and non-bonded interactions (electrostatics, van der Waals forces). Despite their widespread use and continual refinement, traditional force fields possess inherent limitations that impact their predictive power for crucial properties such as conformational energies and interaction strengths. This review establishes a comprehensive baseline of common force fields and their limitations, creating an essential reference point for comparing and validating more computationally intensive quantum chemical methods, including density-functional theory (DFT) and post-Hartree-Fock approaches, in conformational energy research.
Force Field Fundamentals and Evolution
Historical Development and Basic Principles
The conceptual foundation for modern force fields was established with the Consistent Force Field (CFF) in 1968, which introduced a methodology for deriving and validating parameters to describe a wide range of compounds and physical observables. Early protein-specific force fields like ECEPP (Empirical Conformational Energy Program for Peptides) emerged in 1975, relying on crystal data and semi-empirical quantum mechanical (QM) calculations for parameterization. The Allinger force fields (MM1-MM4), developed between 1976-1996, represented another significant advancement, targeting data from electron diffraction, vibrational spectra, heats of formation, and crystal structures.
The basic functional form of potential energy in most molecular mechanics force fields can be expressed as:
[E{\text{total}} = E{\text{bonded}} + E_{\text{nonbonded}}]
where:
[E{\text{bonded}} = E{\text{bond}} + E{\text{angle}} + E{\text{dihedral}}]
and:
[E{\text{nonbonded}} = E{\text{electrostatic}} + E_{\text{van der Waals}}]
This additive approach utilizes simple harmonic potentials for bond and angle deformations, cosine series for dihedral angles, and a combination of Lennard-Jones potentials with Coulomb's law for non-bonded interactions.
The United-Atom to All-Atom Transition
A significant evolution in force field design was the development of united-atom models, which represented nonpolar carbons and their bonded hydrogens as a single particle to enhance computational efficiency. This approach, pioneered with UNICEPP in 1978, could significantly reduce system size since approximately half the atoms in biological macromolecules are hydrogens. While adequate for representing molecular vibrations and bulk properties of small molecules, united-atom force fields demonstrated limitations in accurately treating hydrogen bonds, π-stacking in aromatic systems, and dipole moments when hydrogens were combined with polar heavy atoms. These shortcomings led to a widespread return to all-atom models in force fields like CHARMM22, AMBER ff99, OPLS-AA, and OPLS-AA/L to increase accuracy.
Table 1: Major Biomolecular Force Fields and Their Characteristics
| Force Field | Class | Target Systems | Parameterization Philosophy | Key Strengths |
|---|---|---|---|---|
| AMBER | All-atom, fixed-charge | Proteins, nucleic acids | RESP charges fitted to QM electrostatic potential; focus on structures and non-bonded energies | Accurate description of biomolecular structures |
| CHARMM | All-atom, fixed-charge | Proteins, nucleic acids, lipids | Overestimation of gas-phase dipole moments to implicitly include polarization | Balanced treatment of various biomolecular systems |
| OPLS | All-atom, fixed-charge | Organic molecules, proteins | Optimized for liquid-state properties and thermodynamic observables | Accurate thermodynamic properties |
| GROMOS | United-atom | Biomolecular systems | Parameterized for condensed phase simulations | Computational efficiency for large systems |
| CGenFF | All-atom, fixed-charge | Drug-like molecules | Transferable parameters compatible with CHARMM | Broad coverage of chemical space relevant to drug design |
Current Major Force Fields and Their Limitations
Additive Force Fields in Biomolecular Simulations
The workhorses of contemporary biomolecular simulations include all-atom, fixed-charge force fields such as AMBER, CHARMM, GROMOS, and OPLS. These force fields share a common limitation: the use of fixed atomic charges to model electrostatic interactions, which fails to account for the many-body polarization effects that vary significantly depending on chemical and physical environments. Consequently, non-polarizable force fields cannot capture the conformational dependence of electrostatic properties, which is crucial for accurately modeling phenomena like ligand binding and membrane permeation.
The AMBER force field, first introduced with ff99 in 1999, employs Restrained Electrostatic Potential (RESP) charges derived from quantum mechanical calculations without empirical adjustments. Its philosophy assumes that using these QM-derived charges should necessitate fewer torsional potentials compared to models with empirical charge derivation. The CHARMM force fields utilize a similar approach but typically overestimate gas-phase dipole moments by approximately 20% or more to implicitly account for polarization effects in condensed phases—a recognition that molecular dipole moments are generally larger in condensed phases than in gas phase.
The CHARMM General Force Field (CGenFF) and General AMBER Force Field (GAFF) represent specialized extensions for drug-like molecules. These transferable force fields face the particular challenge of covering vast chemical space with limited parameters, requiring that drug-like molecules be treated as collections of individual chemical group parts whose properties are assumed to remain largely consistent across different molecular environments.
Fundamental Limitations and Systematic Errors
Despite extensive refinement efforts, traditional force fields exhibit several fundamental limitations that constrain their predictive accuracy:
Fixed-Charge Approximation: The atom-centered point charge model cannot describe the anisotropy of charge distribution or account for charge penetration effects (deviation of electrostatic interaction from Coulomb form due to electron shielding when atomic electron clouds overlap). These effects determine equilibrium geometry and energy in molecular complexes. This limitation becomes particularly problematic when molecules transition between environments with different polar characters, such as during ligand binding to proteins or small molecule permeation through membranes.
Lack of Polarization: Electronic polarizability, where electron density adjusts in response to local electric fields, is treated only in a mean-field average way in additive force fields. This simplification fails to capture how charge distribution changes in response to varying local environments, potentially leading to inaccurate representations of intermolecular interactions.
Functional Form Limitations: The simple functional forms of potential energy functions in traditional force fields have been recognized as a major source of inaccuracy. As statistical errors in simulations have decreased with increasing computing power, deficiencies in force fields have become more apparent, manifesting as large deviations in various observables and an inability to accurately predict conformations of proteins and peptides.
Transferability Challenges: Parameters derived from small model compounds may not accurately represent the behavior of larger, more complex systems, particularly in conjugated systems where the properties of chemical groups can vary significantly based on neighboring moieties.
Quantitative Comparison and Experimental Validation
Systematic Force Field Validation Studies
A systematic validation of eight protein force fields against experimental data revealed that while force fields have improved over time, they still exhibit varying performance in describing protein structure and fluctuations. The study, which compared simulation data with experimental NMR measurements and quantified biases toward different secondary structure types, found that recent force field versions provide reasonable descriptions of many structural and dynamical properties but remain imperfect. The research highlighted that deficiencies in force fields became more detectable as statistical errors caused by insufficient sampling were reduced through longer simulation timescales.
Benchmarking with Small Molecule Conformational Analysis
Quantum chemistry calculations using A-value estimation (the free energy difference of cyclohexane conformers) provide a valuable benchmark for evaluating computational methods. Studies comparing calculated A-values with experimental data demonstrate that methods neglecting dispersion forces (like HF) or electron correlation (like standard B3LYP) systematically overestimate A-values, highlighting the significance of properly accounting for these interactions. This benchmark is particularly relevant because A-values depend not only on steric repulsion but also on Baeyer strain, torsional strain, electrostatic interaction, and dispersion forces—all of which must be properly modeled for accurate conformational analysis.
Table 2: Performance of Computational Methods for A-Value Prediction
| Computational Method | Treatment of Dispersion | Typical Accuracy for A-Values | Computational Cost | Key Limitations |
|---|---|---|---|---|
| HF | Neglected | Poor systematic overestimation | Low | No dispersion or electron correlation |
| B3LYP | Indirect, incomplete | Moderate overestimation | Medium-low | Incomplete dispersion treatment |
| B3LYP-D3 | Empirical correction | Good for most substituents | Medium | Empirical parameters |
| M06-2X | Parametrized | Good except for bulky substituents | Medium-high | Overestimation for t-Bu |
| ωB97X-D | Empirical correction | Good overall | Medium-high | Slight overestimation for F |
| MP2 | From wavefunction | Good with adequate basis sets | High | Basis set sensitivity |
| PBE0+MBD | Many-body dispersion | Excellent | High | Computational cost |
Emerging Solutions: Polarizable Force Fields and Advanced Functional Forms
Addressing the Polarization Challenge
To overcome the limitations of fixed-charge models, significant efforts have been directed toward developing polarizable force fields that explicitly treat electronic polarization. These include models based on classical Drude oscillators, fluctuating charges, or induced point dipoles. The Drude polarizable force field, for instance, represents electronic polarization by attaching a charged massless particle (a Drude oscillator) to each atom, connected by a harmonic spring. This approach allows atomic charges to adjust in response to the local electric field, providing a more physical representation of intermolecular interactions.
Simulations of biological systems using polarizable force fields have demonstrated improvements over additive models in multiple contexts, including ion distribution near water-air interfaces, ion permeation through channel proteins, water-lipid bilayer interactions, protein folding, and protein-ligand binding. The explicit treatment of polarization enables more accurate modeling of molecular systems in environments with different polar characters, addressing a fundamental limitation of additive force fields.
Advanced Functional Forms for Improved Physical Representation
Beyond incorporating polarization, next-generation force fields are addressing other limitations in traditional functional forms:
Geometry-Dependent Charge Flux (GDCF): Models that consider how atomic charges change with molecular geometry, implemented in advanced force fields like AMOEBA+(CF), address the assumption of fixed partial charges regardless of local molecular geometry.
Many-Body Dispersion (MBD): Traditional pairwise additive Lennard-Jones potentials fail to capture many-body dispersion effects, which can be significant in larger systems. Incorporation of MBD treatments improves the description of van der Waals interactions.
Sigma-Hole Potentials: Simple solutions to model anisotropic charge distribution in atoms like halogens include attaching off-centered positive charges to represent regions of lower electron density (σ-holes), providing more accurate descriptions of halogen bonding.
Experimental Protocols for Force Field Validation
Conformational Energy Benchmarking
Protocol for A-value comparison of computational methods:
System Selection: Monosubstituted cyclohexanes with diverse substituents (e.g., methyl, tert-butyl, halogen, polar groups) for which experimental A-values are available.
Geometry Optimization: Conformers (equatorial and axial) are optimized using target theoretical methods (e.g., HF, B3LYP, B3LYP-D3, M06-2X, ωB97X-D, MP2) with basis sets such as 6-31G* or LANL2DZ for heavier atoms.
Frequency Calculations: Verification of stationary points as minima (no imaginary frequencies) and calculation of thermal corrections to free energy.
Energy Evaluation: Single-point energy calculations at optimized geometries using higher-level theory (e.g., def2-TZVPD or 6-311+G(2df,2p) basis sets) with solvent correction via Polarizable Continuum Model (PCM) for non-polar substituents.
A-value Calculation: Free energy difference between axial and equatorial conformers: ΔG = Gaxial - Gequatorial.
Statistical Analysis: Comparison with experimental values using correlation coefficients, mean absolute errors, and systematic deviation analysis.
Protein Force Field Validation Protocol
Systematic validation of protein force fields against experimental data:
Folded Protein Simulations: Extensive MD simulations of structured proteins reaching microsecond timescales.
NMR Comparison: Calculation of NMR observables (chemical shifts, J-couplings, residual dipolar couplings) from simulations and comparison with experimental data.
Secondary Structure Propensity: Evaluation of biases toward different secondary structure types by comparing simulation data with experimental data for small peptides that preferentially populate either helical or sheet-like structures.
Folding Simulations: Testing force fields' abilities to fold small proteins (both α-helical and β-sheet structures) to native states.
Error Quantification: Statistical analysis of deviations from experimental data across multiple force fields and systems.
Validation Workflow: Diagram illustrating the systematic protocol for validating force fields against experimental data.
Table 3: Key Computational Resources for Force Field Development and Validation
| Resource Name | Type | Primary Function | Relevance to Force Field Research |
|---|---|---|---|
| GAUSSIAN 09W/16 | Quantum Chemistry Software | Electronic structure calculations | Reference quantum chemical calculations for parameterization and validation |
| CHARMM | Molecular Simulation Package | Biomolecular simulations | Development and application of CHARMM force fields |
| AMBER | Molecular Simulation Package | Biomolecular simulations | Application of AMBER force fields and MD simulation |
| CGenFF Program | Parameterization Tool | Automatic parameter generation | Transferable force field parameter assignment for drug-like molecules |
| AnteChamber | Parameterization Tool | Automatic parameter generation | GAFF and AMBER topology generation |
| CREST | Conformer Sampling Tool | Conformational ensemble generation | Exploration of conformational landscapes for validation |
| Aquamarine (AQM) Dataset | Quantum Chemical Database | Benchmark structures and properties | Validation dataset for large drug-like molecules with solvent effects |
| WebAIM Contrast Checker | Accessibility Tool | Color contrast verification | Ensuring diagram accessibility in publications |
Traditional molecular mechanics force fields provide computationally efficient tools for simulating biomolecular systems but face fundamental limitations in their ability to accurately predict conformational energies and environment-dependent interactions. The fixed-charge approximation, lack of explicit polarization, and simplified functional forms constrain their quantitative accuracy, particularly for properties sensitive to electronic effects. These limitations establish a clear baseline against which quantum mechanical methods must demonstrate superior performance to justify their increased computational cost. As force field development continues to address these challenges through polarizable models and more sophisticated functional forms, the validation benchmarks and protocols outlined here provide critical frameworks for assessing progress. The ongoing refinement of force fields, coupled with emerging quantum chemical approaches and machine learning techniques, promises enhanced accuracy in modeling molecular systems for drug discovery and materials design.
Selecting and Applying Computational Methods: Best-Practice Protocols for DFT and Post-HF
Density Functional Theory (DFT) stands as a cornerstone computational method across chemistry, materials science, and drug development, enabling the prediction of molecular properties from first principles. The accuracy of these predictions, however, is critically dependent on the selection of the exchange-correlation functional [21]. For decades, the B3LYP functional with the 6-31G* basis set has served as a widely used default, particularly in organic chemistry and drug discovery [22]. Yet, as computational chemistry expands into more complex chemical spaces—including transition metal catalysts, surface adsorption, and supramolecular systems—researchers increasingly recognize that no single functional performs universally well across all chemical domains [21] [23]. This reality necessitates a careful, system-specific approach to functional selection grounded in comprehensive validation studies and a deep understanding of functional limitations.
The challenge stems from the fundamental approximations inherent in DFT. While the Hohenberg-Kohn theorem establishes that ground-state properties are uniquely determined by electron density, the exact form of the exchange-correlation functional remains unknown [24]. Accordingly, dozens of functionals have been developed, each with distinct parameterizations and trade-offs between accuracy, computational cost, and applicability. As reaction networks in heterogeneous catalysis and sophisticated drug design place increasing demands on predictive accuracy, the scientific community requires robust guidelines for functional selection that move beyond historical precedent toward evidence-based decision making [21] [24]. This guide provides a comprehensive comparison of DFT functional performance across diverse chemical systems, empowering researchers to make informed choices supported by experimental and high-level computational validation.
Methodological Framework: Benchmarking Approaches and Metrics
Validation Databases and Reference Methods
Assessing functional performance requires comparison against reliable reference data, typically derived from two sources: high-level wavefunction theory and experimental measurements. Coupled Cluster theory with singles, doubles, and perturbative triples (CCSD(T)) is often considered the "gold standard" for molecular energetics, providing reference values for atomization energies, reaction energies, and conformational differences [25] [26]. For transition metal systems with significant static correlation, multireference methods like CASPT2 provide crucial benchmarks, particularly for spin state energies and reaction barriers [23]. Experimental validation includes data from single-crystal adsorption microcalorimetry for surface adsorption enthalpies [21], crystallographic measurements for geometric parameters [27], and spectroscopic data for vibrational frequencies.
Large-scale benchmark studies employ diverse datasets to evaluate functional performance across multiple properties. The Por21 database, for instance, assesses spin state energies and binding properties for iron, manganese, and cobalt porphyrins [23]. Validation sets for organometallic catalysis include transition metal compounds with known crystal structures and thermochemical properties [27]. For surface science, adsorption energies of small molecules on transition metal surfaces provide critical testing grounds [21]. These comprehensive datasets enable statistical evaluation of functional performance across chemical space, moving beyond anecdotal evidence toward quantitative assessment.
Performance Metrics and Statistical Evaluation
Rigorous functional validation employs statistical metrics to quantify performance, including mean unsigned error (MUE), root mean square deviation (RMSD), and linear correlation coefficients (R²) [21] [25]. The "chemical accuracy" target of 1.0 kcal/mol represents the ideal, though this is rarely achieved across diverse systems [23]. Benchmark studies typically report performance against these metrics for multiple properties simultaneously—geometries, energies, and frequencies—as a functional may excel in one area while failing in another [26].
Statistical analysis reveals whether functional errors are systematic or random, informing correction strategies. Some studies employ percentile ranking or grading systems to facilitate functional selection [23]. For instance, a grade "A" functional might fall in the top 20th percentile for a specific dataset, while grade "F" functionals perform unacceptably poorly. This categorical approach helps researchers quickly identify promising candidates for their specific systems while avoiding known pitfalls.
Comparative Performance Across Chemical Systems
Surface Science and Heterogeneous Catalysis
The adsorption of atoms and small molecules on transition metal surfaces represents a critical test for functionals in heterogeneous catalysis. A 2020 benchmark study evaluated multiple functionals for describing adsorption of CH₃I, CH₃, I, H, and CH₄ dissociation on Ni(111) compared to single-crystal adsorption microcalorimetry experiments [21]. The results demonstrated significant functional dependence, with no single functional achieving quantitative accuracy (within experimental error) for all adsorption processes studied. However, PBE-D3 and RPBE-D3 emerged as the most consistently accurate when allowing for a ±20 kJ/mol error margin [21].
Table 1: Performance of Select Functionals for Adsorption on Ni(111)
| Functional | CH₃I Molecular Adsorption | CH₃ Adsorption | I Adsorption | H Adsorption | CH₄ Dissociative Adsorption |
|---|---|---|---|---|---|
| PBE-D3 | Accurate within ±20 kJ/mol | Accurate | Accurate | Accurate | Accurate |
| RPBE-D3 | Accurate within ±20 kJ/mol | Accurate | Accurate | Accurate | Accurate |
| optB88-vdW | Most accurate | Less accurate | Less accurate | Less accurate | Less accurate |
| BEEF-vdW | Less accurate | Most accurate | Less accurate | Less accurate | Less accurate |
| SCAN | Less accurate | Less accurate | Most accurate | Less accurate | Less accurate |
| SCAN-RVV10 | Less accurate | Less accurate | Less accurate | Most accurate | Less accurate |
The study highlights that quantitative agreement between DFT and experiment is both system- and functional-dependent, emphasizing the need for continued experimental benchmarking [21]. For surface chemistry applications, dispersion-corrected GGA functionals like PBE-D3 and RPBE-D3 provide the most consistent starting point, though system-specific validation remains essential.
Transition Metal Chemistry and Organometallic Catalysis
Transition metal systems present particular challenges due to complex electronic structures, multireference character, and delicate spin state balances. A comprehensive 2023 benchmark evaluated 250 electronic structure methods for spin states and binding properties of iron, manganese, and cobalt porphyrins [23]. The results revealed that current approximations fail to achieve chemical accuracy by a significant margin, with the best-performing methods achieving MUEs of ~15 kcal/mol—far from the 1 kcal/mol target.
Table 2: Top-Performing Functionals for Transition Metal Porphyrins (Por21 Database)
| Functional | Grade | Type | MUE (kcal/mol) | Spin State Performance | Binding Energy Performance |
|---|---|---|---|---|---|
| GAM | A | GGA | <15.0 | Excellent | Excellent |
| revM06-L | A | meta-GGA | <15.0 | Excellent | Excellent |
| M06-L | A | meta-GGA | <15.0 | Excellent | Excellent |
| r2SCAN | A | meta-GGA | <15.0 | Excellent | Excellent |
| r2SCANh | A | Hybrid meta-GGA | <15.0 | Excellent | Excellent |
| HCTH | A | GGA | <15.0 | Excellent | Excellent |
| B3LYP | C | Hybrid GGA | ~20-23 | Moderate | Moderate |
| B2PLYP | F | Double-hybrid | >30 | Poor | Poor |
Notably, local functionals (GGAs and meta-GGAs) generally outperformed hybrid and double-hybrid functionals for these challenging transition metal systems [23]. Functionals with high percentages of exact exchange, including range-separated and double-hybrid functionals, often exhibited catastrophic failures for spin state energetics. This finding contradicts the conventional wisdom that exact exchange improves treatment of static correlation, highlighting the specialized challenges of transition metal applications.
For geometric properties of transition metal compounds, a 2012 validation study of catalysts for olefin metathesis and other homogeneous reactions found that dispersion-corrected functionals like B97D, wB97XD, and M06 provided the most accurate structures [27]. The study emphasized that dispersion corrections are essential for accurate geometries, particularly for weakly interacting systems prevalent in organometallic chemistry.
Main-Group Molecules and Non-Covalent Interactions
For main-group molecules and non-covalent interactions, modern range-separated and dispersion-corrected functionals typically deliver superior performance. A study on beryllium, tungsten, and hydrogen-containing molecules identified ωB97XD as the top performer for atomization energies, closely followed by B97D, M06, B3LYP, and M11 [26]. For vibrational frequencies, HSEH1PBE, B3LYP, and M11 showed the best accuracy, while M11, ωB97XD, and HSEH1PBE excelled for bond lengths [26].
The importance of dispersion corrections emerges consistently across studies. For example, in the r2SCAN functional series, the dispersion-corrected r2SCAN-D4 variant significantly outperformed its uncorrected counterpart for non-covalent interactions [23]. Similarly, in crystal structure prediction—where dispersion forces dominate packing arrangements—the r2SCAN-D3 functional enables highly accurate lattice energy rankings [28].
Specialized Applications: From Drug Development to Energy Materials
Pharmaceutical Development and Biomolecular Systems
In pharmaceutical development, DFT guides drug design through molecular electrostatic potential maps, Fukui functions, and reaction site identification [24]. The B3LYP functional with 6-31G(d,p) basis set remains widely employed for these applications due to its balanced performance and computational efficiency [22]. For drug formulation design, DFT elucidates API-excipient interactions, co-crystal formation, and nanocarrier optimization [24]. Recent advances integrate DFT with molecular mechanics in multiscale frameworks like ONIOM, enabling accurate modeling of drug-receptor interactions while managing computational cost [24].
For protein energy landscapes—relevant to aggregation propensity and misfolding diseases—DFT provides insights into conformational dynamics, though classical force fields typically handle larger-scale simulations [29] [30]. Experimental studies using nanoaperture optical tweezers have directly resolved energy landscapes of single proteins like bovine serum albumin, revealing three-state conformation dynamics with temperature [30]. These experimental benchmarks could inform future DFT development for biomolecular systems.
Information-Theoretic Approach for Correlation Energies
Beyond traditional DFT, the information-theoretic approach (ITA) offers promising alternatives for predicting electron correlation energies. Using descriptors derived from Hartree-Fock electron density—including Shannon entropy, Fisher information, and relative Rényi entropy—linear regression models can predict post-Hartree-Fock correlation energies at significantly reduced computational cost [25]. This approach has demonstrated chemical accuracy for diverse systems including octane isomers, polymeric structures, and molecular clusters [25].
The LR(ITA) protocol achieves RMS deviations of <2.0 mH for octane isomers and ~1.5-4.0 mH for linear polymers, rivaling the accuracy of more expensive embedded fragmentation methods like the generalized energy-based fragmentation (GEBF) approach [25]. While not replacing conventional DFT, ITA quantities provide computationally efficient descriptors for correlation energy prediction, particularly in large systems where post-Hartree-Fock calculations become prohibitive.
Functional Selection Workflow and Recommendations
Based on comprehensive benchmarking data, we propose a systematic workflow for functional selection (illustrated above) with the following specific recommendations:
For transition metal systems (particularly spin states and binding energies): Prefer local functionals like GAM, revM06-L, M06-L, or r2SCAN, which outperform hybrids for these challenging applications [23]. Avoid double-hybrids and high-exact-exchange functionals.
For non-covalent interactions and molecular clusters: Select dispersion-corrected functionals like ωB97XD, B97D, or r2SCAN-D3 [25] [26]. Dispersion corrections are essential for accurate binding energies and geometries.
For surface science and adsorption phenomena: Use PBE-D3 or RPBE-D3, which provide the most consistent performance across various adsorption systems [21].
For main-group thermochemistry and organic molecules: B3LYP remains reasonable, though ωB97XD, M11, and HSEH1PBE often provide superior accuracy [26].
For geometric properties: M11, ωB97XD, and HSEH1PBE deliver excellent bond lengths, while dispersion corrections are critical for non-covalent interactions [26] [27].
Regardless of the initial selection, system-specific validation against experimental data or high-level reference calculations remains essential [21]. When possible, test multiple functionals from different classes to quantify uncertainty in predictions.
Experimental Protocols for Functional Validation
Adsorption Calorimetry Benchmarking
Single-crystal adsorption microcalorimetry provides experimental benchmarks for surface science applications [21]. The protocol involves:
- Preparing well-defined single crystal surfaces under ultra-high vacuum
- Measuring heats of adsorption via microcalorimetry at controlled temperatures (e.g., 160 K)
- Comparing DFT-calculated adsorption enthalpies with experimental values
- Evaluating functional performance using statistical metrics (MUE, RMSD)
This approach revealed that while each adsorption process studied on Ni(111) had at least one accurately describing functional, none performed quantitatively well across all systems [21].
High-Level Wavefunction Theory Benchmarks
For molecular systems, CCSD(T) with complete basis set extrapolation provides reference energies [26]. The standard protocol includes:
- Geometry optimization at CCSD(T)/cc-pVQZ level
- Energy evaluation with cc-pVQZ/cc-pV5Z extrapolation to CBS limit
- Accounting for core correlation effects (3-5% for heavy elements)
- Comparing DFT results against these reference values
For transition metals with potential multireference character, CASPT2 references provide crucial validation, though reference quality must be critically assessed [23].
Research Reagent Solutions: Computational Tools
Table 3: Essential Computational Tools for DFT Validation Studies
| Tool Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| Quantum Chemistry Software | VASP [21], Gaussian, ORCA | DFT calculations with various functionals | Production calculations across chemical systems |
| Wavefunction Theory Codes | MOLPRO, CFOUR, MRCC | High-level reference calculations (CCSD(T), CASPT2) | Generating benchmark data for validation |
| Analysis Tools | Multiwfn, IGMH, NCIPLOT | Electron density analysis, non-covalent interaction visualization | Information-theoretic approach, bonding analysis |
| Database Resources | Por21 database [23], GMTKN55, NIST CCCBDB | Curated benchmark datasets | Functional testing and validation |
| Crystal Structure Prediction | r2SCAN-D3 method [28] | Polymorph prediction and ranking | Pharmaceutical development, materials design |
The functional landscape beyond B3LYP/6-31G* is rich and varied, with specialized performers emerging for different chemical domains. No single functional currently achieves universal chemical accuracy, necessitating careful, system-aware selection. Key trends include the superiority of local functionals for challenging transition metal systems, the critical importance of dispersion corrections for non-covalent interactions, and the consistent performance of PBE-D3/RPBE-D3 for surface science.
Future developments will likely focus on machine-learning-augmented functionals, systematic uncertainty quantification, and efficient implementations of high-performing functionals like r2SCAN [24]. The information-theoretic approach offers promising alternatives for correlation energy prediction [25], while multiscale frameworks integrate DFT with molecular mechanics for complex systems [24]. As these methods mature, functional selection may become more systematic and less empirical. Until then, the guidelines presented here—grounded in comprehensive benchmark studies—provide researchers with evidence-based strategies for navigating the complex functional landscape, ultimately enhancing the reliability of computational predictions across chemistry, materials science, and drug development.
Basis Set Selection and the Role of Composite Methods for Efficiency and Accuracy
Computational chemistry, like all attempts to simulate reality, is defined by tradeoffs. Reality is far too complex to simulate perfectly, and scientists have developed a plethora of approximations, each of which reduces both the cost and the accuracy of the simulation [31]. The responsibility of the researcher is to choose an appropriate method that best balances speed and accuracy for the task at hand. This balance can be framed in terms of Pareto optimality, where a "frontier" exists of optimal speed/accuracy combinations, with suboptimal combinations being inefficient [31]. The choice of basis set—the set of mathematical functions used to represent molecular orbitals—is one of the most critical approximations influencing this balance. It heavily influences the accuracy, CPU time, and memory usage of a calculation [32]. Simultaneously, the development of composite methods represents a strategic approach to accessing high accuracy without prohibitive computational cost by combining results from several calculations using different levels of theory and basis sets [33]. This guide provides a comparative analysis of basis set selection strategies and composite methods, framed within the context of validating conformational energies and other properties in Density Functional Theory (DFT) and post-Hartree-Fock (post-HF) research.
Basis Set Fundamentals and Hierarchy
A basis set in quantum chemistry consists of a set of basis functions (atomic orbitals) used to build molecular orbitals. The fundamental trade-off is that a larger, more complete basis set typically yields a more accurate result but at a significantly higher computational cost [32].
Standard Basis Set Types and Nomenclature
Basis sets are systematically improved by increasing both the number of basis functions per atom and their flexibility. The standard hierarchy, from smallest/least accurate to largest/most accurate, is summarized in the table below [32].
Table 1: Standard Basis Set Hierarchy and Typical Performance Characteristics
| Basis Set | Description | Typical Use Case | Computational Cost (Relative to SZ) | Energy Error (Example) [eV] |
|---|---|---|---|---|
| SZ | Single Zeta | Minimal basis; quick test calculations [32] | 1 (Reference) | 1.8 [32] |
| DZ | Double Zeta | Pre-optimization of structures [32] | 1.5 | 0.46 [32] |
| DZP | Double Zeta + Polarization | Geometry optimizations of organic systems [32] | 2.5 | 0.16 [32] |
| TZP | Triple Zeta + Polarization | Recommended default for best performance/accuracy balance [32] | 3.8 | 0.048 [32] |
| TZ2P | Triple Zeta + Double Polarization | Accurate results; good description of virtual space [32] | 6.1 | 0.016 [32] |
| QZ4P | Quadruple Zeta + Quadruple Polarization | Benchmarking [32] | 14.3 | Reference [32] |
Polarization functions are crucial for describing the deformation of electron clouds when bonds form, while diffuse functions (often denoted with "++" or "aug-") are important for anions and weak interactions like hydrogen bonding [34] [33].
Quantitative Performance of Basis Sets
The impact of basis set selection on accuracy and cost can be quantified. For instance, in a study on a carbon nanotube, the energy error per atom decreased dramatically from 1.8 eV with an SZ basis set to 0.016 eV with a TZ2P basis, but the computational cost increased by a factor of 14 when moving to QZ4P [32]. It is critical to note that errors in absolute energies are often systematic and can partially cancel when calculating energy differences, such as conformational energies, reaction barriers, or binding energies. Therefore, a smaller basis like DZP might yield excellent results for energy differences even though its absolute energy error is relatively large [32].
Composite Methods: Strategic Combinations for High Accuracy
Composite methods aim for high accuracy, often to within 1 kcal/mol of experimental values (chemical accuracy), by combining the results of several calculations. They strategically use high-level theory with small basis sets and lower-level theory with large basis sets, relying on error cancellation [33].
Gaussian-n Theories
The Gaussian-n theories (G1, G2, G3, G4) were the first systematic composite models with broad applicability [33].
- G2 Theory: This method uses a multi-step procedure. The geometry is optimized at the MP2/6-31G(d) level. Subsequently, single-point energy calculations are performed at higher levels, including QCISD(T)/6-311G(d) and MP4 with larger basis sets to account for polarization and diffuse functions. The final energy includes an empirical "Higher Level Correction" (HLC) and a scaled zero-point vibrational energy [33].
- G3/G4 Theories: These are evolutions of G2. G3 uses smaller basis sets (6-31G) for some steps and a larger one (G3large) for the final MP2 calculation, also incorporating a spin-orbit correction. G4 introduces further refinements, such as extrapolation to the Hartree-Fock basis set limit, using CCSD(T) instead of QCISD(T) for the highest-level calculation, and employing B3LYP for geometries and thermochemical corrections [33].
Table 2: Comparison of Selected Composite Methods
| Method | Core Philosophy | Key Components | Target Accuracy | Applicability |
|---|---|---|---|---|
| Gaussian-4 (G4) | Fixed recipe with empirical correction [33] | B3LYP geometries, CCSD(T) energy, large MP2 basis set, HLC [33] | ~1 kcal/mol for thermochemistry [33] | Main group elements, transition metals [33] |
| ccCA | Non-empirical, uses Dunning's basis sets [33] | MP2/CBS reference, ΔCCSD(T) correction, core-valence, relativistic, ZPVE corrections [33] | High accuracy without fitted parameters [33] | Main group elements [33] |
| Feller-Peterson-Dixon (FPD) | Flexible, system-dependent sequence [33] | CCSD(T) with very large basis sets (up to aug-cc-pV8Z), extrapolation to CBS, core/valence & relativistic corrections [33] | ~0.3 kcal/mol RMS error (benchmarked) [33] | Small systems (≤10 first/second row atoms) [33] |
| B97-3c | Simplified DFT composite [31] | B97 functional, modified def2-TZVP basis (mTZVP), D3 dispersion, SRB correction [31] | Benchmark-accuracy for key properties [31] | Large systems, general purpose [31] |
| r²SCAN-3c | Modern meta-GGA composite [31] | r²SCAN functional, modified TZVP basis, D4 dispersion, gCP correction [31] | High accuracy and robustness [31] | Large-scale screenings [31] |
Grimme's "3c" Methods for Efficiency
A distinct class of composite methods, pioneered by Grimme, is designed for high efficiency on large systems. These methods use a reduced-cost base method (e.g., HF or DFT with a small basis set) and layer on physically motivated corrections to recover accuracy [31].
- HF-3c: Uses Hartree-Fock with a minimal basis set (MINIS), corrected with D3 dispersion, a geometric counterpoise (gCP) correction for basis set incompleteness error, and a short-range basis (SRB) correction. It is surprisingly good for geometry optimization and non-covalent interactions [31].
- PBEh-3c: A DFT-based composite method using a modified polarized double-zeta basis set (def2-mSVP), the D3 and gCP corrections, and a reparameterized PBEh functional [31].
- B97-3c and r²SCAN-3c: These represent more advanced DFT composites. B97-3c uses a modified triple-zeta basis (mTZVP) and the D3 and SRB corrections, often outperforming standard DFT with much larger basis sets. The r²SCAN-3c method is noted for its robustness and benchmark accuracy, drastically shifting the balance between efficiency and accuracy for large systems [31].
Experimental Protocols for Validation
Validating the performance of basis sets and composite methods requires comparison against reliable benchmark data, which can be experimental results or highly accurate theoretical values.
Protocol for Energetic Properties
A robust protocol for validating conformational energies, atomization energies, or interaction energies involves:
- Reference Data Selection: Use a well-established benchmark set, such as the GMTKN55 database for general main-group thermochemistry, kinetics, and non-covalent interactions, or the S66 set for non-covalent interactions [31].
- Geometry Optimization: Optimize all molecular structures for each benchmark system using a consistent, reasonably accurate method. Many composite methods like G4 and ccCA use B3LYP/6-31G(2df,p) or similar for this step [33].
- Single-Point Energy Calculations: Calculate the single-point energies for the optimized structures using the method/basis set combination being tested. For composite methods, this involves the specific sequence of calculations defined by the method [33].
- Error Calculation: For each benchmark entry, calculate the error as: Error = E_{calculated} - E_{reference}. Then, compute statistical measures like Mean Absolute Error (MAE) and Root-Mean-Square Error (RMSE) across the entire dataset [35] [31].
- Statistical Analysis: Compare the MAE and RMSE of different methods. A method achieving an MAE below 1 kcal/mol is generally considered to have reached chemical accuracy for energetic properties [33].
Protocol for Structural Properties
Validating the accuracy of calculated molecular structures, such as bond lengths, follows a similar approach:
- Reference Structures: Use a set of molecules with high-resolution gas-phase experimental equilibrium structures (e.g., from microwave spectroscopy or electron diffraction) or highly accurate theoretical re values [36].
- Geometry Optimization: Perform a full geometry optimization for each molecule in the test set using the method and basis set under investigation.
- Linear Regression: Perform a linear regression of calculated bond lengths versus experimental/benchmark values. The slope, intercept, and correlation coefficient (R²) are key indicators of performance [36].
- Statistical Analysis: Calculate the MAE and RMSE for the bond lengths. For example, high-level composite methods like the FPD approach can achieve RMS deviations for equilibrium bond lengths of 0.0020 Å for heavy atoms [33].
The following diagram illustrates a generalized validation workflow for computational methods.
Successful computational research relies on a suite of software, databases, and computational resources. The table below details key "research reagent solutions" for studies involving basis set selection and composite methods.
Table 3: Essential Research Reagents and Resources for Computational Studies
| Resource / Tool | Type | Primary Function | Relevance to Validation Studies |
|---|---|---|---|
| GMTKN55 Database [31] | Benchmark Database | A diverse collection of 55 benchmark sets for testing general main-group methods. | The primary standard for validating the accuracy of DFT and composite methods for thermochemistry, kinetics, and non-covalent interactions. |
| GAUSSIAN, ORCA, Q-Chem | Quantum Chemistry Software | Suites that implement a wide range of quantum chemical methods and basis sets. | Used to perform geometry optimizations, single-point energy calculations, and automated composite method computations (e.g., G4). |
| B97-3c / r²SCAN-3c [31] | Composite Method | Efficient, all-in-one methods for geometry optimization and energy calculation. | Used as a production method for large systems or as a high-quality, cost-effective reference for method comparison. |
| Dunning's cc-pVXZ [34] [33] | Basis Set Family | A systematically improvable series of basis sets for high-accuracy calculations. | Essential for studies approaching the complete basis set (CBS) limit and as components in composite methods like ccCA. |
| Counterpoise (CP) Correction [34] | Computational Protocol | Corrects for Basis Set Superposition Error (BSSE). | Critical for obtaining accurate interaction energies (e.g., conformational energies, binding affinities) with small to medium basis sets. |
| CHEMBL, ZINC [37] [38] | Chemical Database | Publicly available databases of molecules with annotated bioactivity or commercial availability. | Sources for real-world molecular structures to test method performance on pharmaceutically relevant compounds. |
The selection of an appropriate basis set or composite method is not a one-size-fits-all decision but a strategic choice dictated by the specific property of interest, the system size, and the available computational resources. For routine studies on large systems, modern composite methods like B97-3c or r²SCAN-3c offer an exceptional balance of speed and accuracy [31]. When the highest possible accuracy is required for smaller systems, sophisticated composite approaches like G4 or ccCA are the tools of choice [33]. The fundamental principle across all scenarios is that validation against robust benchmark data is indispensable. By leveraging the hierarchies and protocols outlined in this guide, researchers can make informed decisions that streamline the drug discovery process and enhance the reliability of computational predictions in DFT and post-HF research.
The accurate prediction of molecular conformation and its associated energy is a cornerstone of computational chemistry, with profound implications for drug design and materials science. The challenge lies not only in sampling the vast conformational space but also in evaluating the energies of these conformers with high accuracy. No single computational method optimally balances efficiency and accuracy across both tasks. Consequently, modern research increasingly relies on multi-level workflows that integrate rapid conformational sampling methods with high-precision single-point energy calculations. This guide objectively compares the performance of prevalent methods and workflows, providing a structured overview of their protocols, accuracy, and integration strategies for researchers and drug development professionals.
Performance Benchmarking of Computational Methods
Comparative Accuracy of Conformational Energies
The performance of computational methods is most meaningfully assessed by benchmarking against highly accurate reference data, such as that provided by DLPNO-CCSD(T) theory, often considered the "gold standard" for single-point energies [39] [40]. A comprehensive study evaluated 158 conformational energies of 145 organic molecules, which are model fragments commonly found in drug-like molecules, against DLPNO-CCSD(T) reference values [40].
Table 1: Mean Absolute Errors (kcal mol⁻¹) for Conformational Energies vs. DLPNO-CCSD(T) Benchmark [40]
| Method Category | Specific Method | Mean Absolute Error (MAE) |
|---|---|---|
| Wavefunction Theory | MP2 | 0.35 |
| Density Functional Theory | B3LYP | 0.69 |
| Wavefunction Theory | HF | 0.81 - 1.00 |
| Molecular Mechanics (Force Fields) | MM3-00 | 1.28 |
| Molecular Mechanics (Force Fields) | MMFF94 | 1.30 |
| Molecular Mechanics (Force Fields) | MMX | 1.77 |
| Molecular Mechanics (Force Fields) | MM+ | 2.01 |
| Molecular Mechanics (Force Fields) | DREIDING | 3.63 |
| Molecular Mechanics (Force Fields) | UFF | 3.77 |
The data reveals a clear hierarchy of accuracy. MP2 demonstrated the lowest mean error, followed closely by B3LYP and Hartree-Fock [40]. Among force fields, MM3-00 and MMFF94 showed the best performance, while more generic force fields like UFF and DREIDING exhibited significantly larger errors [40]. This underscores the importance of method selection for accurate energy ranking.
The Critical Role of Method Selection in Energy Evaluation
The choice of method for single-point energy calculations is critical and depends on the desired balance between computational cost and accuracy.
- Coupled Cluster Theory: Methods like DLPNO-CCSD(T) provide the highest accuracy and are considered the gold standard for final energy assessments, but they are computationally demanding [39] [40].
- Double-Hybrid DFT (DH-DFT): Functionals like B2PLYP and revDSD-PBEP86-D4 incorporate both HF exchange and MP2 correlation, offering improved accuracy over standard DFT, but typically require larger basis sets [39].
- Standard DFT and HF: While B3LYP is widely used, it is known to suffer from inherent errors like missing London dispersion effects, which can be mitigated using modern dispersion corrections (e.g., D3, D4) and composite methods [39] [16]. Hartree-Fock theory is generally inferior for quantitative energy predictions but forms the basis for more advanced post-HF methods [41].
Integrated Workflow Protocols
Automated workflows streamline the process from conformational sampling to final energy evaluation, reducing user intervention and potential errors.
The XTBDFT Workflow
The XTBDFT workflow exemplifies a multi-level approach, leveraging the speed of semi-empirical methods for sampling and the accuracy of DFT for final evaluation [42].
Diagram 1: XTBDFT multi-level workflow
This workflow begins with a comprehensive conformational search using GFN-XTB, a semi-empirical method that is fast and effective for organic and some metal-organic systems [42]. Promising low-energy conformers identified from this search are then passed to a higher-level DFT method for a final, accurate single-point energy calculation [42]. This protocol efficiently narrows down thousands of potential conformers to a manageable set for high-cost DFT computation.
The PyConSolv Workflow for Explicit Solvent Environments
For systems where explicit solvent effects are critical, the PyConSolv package provides an automated workflow using classical molecular dynamics (MD) for sampling [43].
Diagram 2: PyConSolv automated conformer generation
This protocol is particularly valuable for metal-containing complexes and systems where solvent can induce significant conformational changes [43]. The key steps involve:
- Automated Parametrization: Generating force field parameters for the system, including metals, using QM calculations (e.g., with ORCA) [43].
- Explicit Solvent MD Sampling: Running lengthy MD simulations in explicit solvent to sample the conformational landscape [43].
- Clustering and QM Refinement: Clustering the MD trajectory to identify representative conformers, which are then ranked using high-level QM single-point energy calculations [43].
This approach combines the realistic solvation environment of classical MD with the accuracy of QM energy evaluation.
Detailed Experimental Protocols
- Conformer Sampling: Execute a conformational search using CREST with the GFN-xTB Hamiltonian. This typically involves a series of meta-dynamics simulations and geometry optimizations.
- Initial Filtering: Collect all unique conformers generated by CREST.
- Geometry Optimization (Optional): For the most promising conformers, a re-optimization can be performed using a more accurate method (e.g., a DFT functional like B3LYP or a better semi-empirical method) with an appropriate basis set.
- Final Single-Point Energy Calculation: Perform a high-level single-point energy calculation (e.g., using double-hybrid DFT or DLPNO-CCSD(T)) on the optimized geometries to obtain accurate relative energies.
- Analysis: Rank the conformers based on the final single-point energies.
This protocol adapts the double decoupling method (DDM) for use with an implicit solvation model, enhancing sampling efficiency.
- End-State Sampling: Perform extensive conformational sampling (e.g., using Temperature Replica Exchange MD - TREMD) for the bound (host-guest complex) and unbound (free ligand and host) states in implicit solvent.
- Conformational Restraining: Apply harmonic distance restraints between atoms within 6 Å in the binding site to limit conformational space in intermediate states.
- Alchemical Transformation: Calculate the free energy cost of decoupling the ligand from its environment through a series of intermediate states where ligand partial charges and van der Waals interactions are scaled to zero. This is performed with both conformational and Boresch-style orientational restraints applied.
- Free Energy Calculation: Use free energy perturbation (FEP) or thermodynamic integration (TI) to compute the free energy change over the alchemical pathway.
- Restraint Corrections: Analytically calculate the free energy cost of releasing the applied restraints to obtain the absolute binding free energy.
The Scientist's Toolkit: Essential Research Reagents and Software
Table 2: Key Software and Computational Tools for Workflow Integration
| Tool Name | Primary Function | Role in Workflow | Key Features |
|---|---|---|---|
| CREST/GFN-xTB [42] [43] | Conformer Sampling | Initial Stage | Rapid, semi-empirical sampling of conformational space. |
| ORCA [39] [43] | Quantum Chemistry | Energy Evaluation | High-accuracy single-point energies (DFT, MP2, DLPNO-CCSD(T)). |
| PyConSolv [43] | Automated Workflow | Integration & Sampling | Automated parametrization and explicit solvent MD for conformer generation. |
| AmberTools [43] | MD Simulation | Sampling & Parametrization | Force field parametrization, MD setup, and trajectory analysis. |
| MultiWFN [43] | Wavefunction Analysis | Parametrization | Calculation of RESP charges for force field development. |
| GoodVibes [42] | Data Analysis | Final Analysis | Thermochemical and vibrational analysis of QM results. |
| XTBDFT [42] | Automated Workflow | End-to-End Integration | Scripted pipeline from GFN-XTB sampling to DFT energy refinement. |
The integration of conformational sampling and single-point energy calculations represents a powerful paradigm in computational chemistry. Benchmarking studies consistently show that while force fields like MMFF94 and MM3-00 are useful for initial screening, high-accuracy conformational energies require post-HF methods like MP2 or DLPNO-CCSD(T) or carefully parameterized double-hybrid DFT [39] [40]. The emergence of automated workflows like XTBDFT and PyConSolv demonstrates the practical implementation of multi-level strategies, leveraging the strengths of different computational methods to achieve both breadth in sampling and depth in accuracy. For researchers in drug development, these integrated protocols provide a robust framework for predicting molecular properties with confidence, ultimately accelerating the design and optimization of new therapeutic agents.
Within computer-aided drug design, the accurate calculation of conformational energies—the relative energies of different three-dimensional shapes a molecule can adopt—is a cornerstone of reliable in-silico predictions. The stability and biological activity of a drug candidate are intimately tied to its low-energy conformations [9]. Researchers therefore depend on computational methods that can correctly rank these conformers. This case study examines the performance of various computational chemistry methods, from force fields to high-level quantum mechanical (QM) approaches, for this critical task. The analysis is framed within the broader thesis of validating computational methods against highly accurate "gold-standard" quantum chemistry references, specifically focusing on their application to model organic fragments commonly found in drug-like molecules [9].
Reference Data and Benchmarking Molecules
The foundation of any validation study is a robust benchmark set. Recent research has established a curated set of 145 reference organic molecules specifically selected to represent model fragments encountered in drug design [9]. This set includes a diverse range of chemical functionalities: hydrocarbons, haloalkanes, conjugated compounds, and oxygen-, nitrogen-, phosphorus-, and sulphur-containing molecules [9]. For these molecules, 158 conformational energies and barriers were calculated using a wide array of methods.
The reference values against which all methods were benchmarked were computed at the DLPNO-CCSD(T) level of theory [9]. This coupled cluster method is considered a "gold standard" in quantum chemistry for its high accuracy, providing a reliable benchmark for evaluating the performance of more computationally efficient methods [9].
Computational Methods Workflow
The general workflow for calculating and validating conformational energies involves generating an ensemble of possible conformers for a molecule and then calculating their relative energies using different levels of theory. The following workflow diagram illustrates a modern, efficient multi-level approach for generating high-quality conformational ensembles, which can then be used for energy benchmarking.
Diagram 1: Efficient workflow for generating DFT-quality conformational ensembles [44] [45] [4].
Performance Comparison of Computational Methods
Quantitative Accuracy Against DLPNO-CCSD(T) Reference
The performance of various computational methods was quantified by their mean absolute error (MAE) relative to the DLPNO-CCSD(T) reference energies for the benchmark set of organic molecules [9]. The results are summarized in the table below.
Table 1: Performance of computational methods for calculating conformational energies. MAE is Mean Absolute Error relative to DLPNO-CCSD(T)/cc-pVTZ reference [9].
| Method Category | Specific Method | Mean Absolute Error (MAE, kcal mol⁻¹) | Key Characteristics |
|---|---|---|---|
| High-Level QM | MP2 | 0.35 | High accuracy, computationally expensive |
| B3LYP | 0.69 | Popular DFT functional, good balance | |
| Hartree-Fock (HF) | 0.81 - 1.00 | Low computational cost, moderate accuracy | |
| Force Fields | MMFF94 | 1.30 | Designed for drug-like molecules |
| MM3-00 | 1.28 | Allinger's force field, high accuracy | |
| MM3-96 | 1.40 | Allinger's force field | |
| MMX | 1.77 | MM2-91 clone | |
| MM+ | 2.01 | MM2-91 clone | |
| MM4 | 2.05 | Allinger's force field | |
| DREIDING | 3.63 | Generic force field | |
| UFF | 3.77 | Universal Force Field |
Analysis of Performance Data
The benchmark data reveals a clear hierarchy of accuracy. Among the quantum mechanical methods, MP2 demonstrated the lowest error (0.35 kcal mol⁻¹), making it an excellent choice for high-accuracy studies, though its computational cost can be prohibitive for very large systems [9]. The density functional theory (DFT) functional B3LYP also performed well (0.69 kcal mol⁻¹), offering a good balance between accuracy and computational cost [9].
Among molecular mechanics force fields, which are crucial for high-throughput screening due to their speed, MM3-00 and MMFF94 were the top performers, with MAEs of approximately 1.3 kcal mol⁻¹ [9]. The MM2 clones (MMX, MM+) and MM4 showed intermediate performance, while the generic force fields UFF and DREIDING had the highest errors (over 3.6 kcal mol⁻¹) and are not recommended for accurate conformational energy ranking in drug fragments [9]. An error of 1 kcal mol⁻¹ is significant as it is comparable to the thermal energy (RT) at room temperature, and can thus lead to incorrect predictions of the predominant conformer in an ensemble.
Detailed Experimental Protocols
Protocol 1: High-Accuracy Benchmarking with DLPNO-CCSD(T)
This protocol outlines the steps used to generate the benchmark data against which other methods are validated [9].
- System Selection: Curate a set of 145 small organic molecules representing common fragments in drug-like molecules.
- Conformer Generation: For each molecule, generate an ensemble of low-energy conformers using a robust sampling algorithm.
- Geometry Optimization: Optimize the geometry of each conformer using a high-level DFT method (e.g., ωB97X-D/def2-SVP).
- Reference Energy Calculation: Perform a single-point energy calculation on the optimized geometry at the DLPNO-CCSD(T)/cc-pVTZ level of theory. This serves as the reference energy.
- Method Benchmarking: Calculate the conformational energies (energy differences between conformers) using the methods under investigation (e.g., other DFT functionals, force fields).
- Error Calculation: Compute the mean absolute error (MAE) for each method by comparing its conformational energies to the DLPNO-CCSD(T) references.
Protocol 2: Efficient Multi-Level Workflow for Drug Fragments
For practical applications on larger systems, a multi-level workflow that balances accuracy and computational cost is essential. The following protocol, adapted from recent literature, is highly effective for generating DFT-quality conformational ensembles for drug fragments and their dimers [44] [45] [4].
- Initial Ensemble Generation: Use the Conformer-Rotamer Ensemble Sampling Tool (CREST) with its underlying GFN2-xTB semi-empirical method to generate a broad, initial set of conformers [44] [4].
- Pre-screening and Reoptimization: Reoptimize every conformer from step 1 using a fast, composite DFT method like B97-3c. This method was selected for its excellent performance in screening studies on model dimers [44] [45].
- Curate Ensemble: Discard duplicate conformers (based on a root-mean-square deviation, RMSD, threshold) and any conformers with unrealistically high energies [44] [4].
- High-Level DFT Optimization: Reoptimize the remaining, unique conformers using a more robust DFT functional and basis set, such as ωB97X-D4/def2-SVP [44] [45].
- Final Energetics: Perform a vibrational frequency calculation to confirm the structure is a minimum (not a transition state) and to obtain thermodynamic corrections. Finally, compute a ultra-fine single-point energy using a large basis set like ωB97X-V/def2-QZVPP for the most accurate final energies [44] [45].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential computational tools and resources for conformational energy calculations.
| Tool/Resource Name | Type/Category | Primary Function in Research |
|---|---|---|
| DLPNO-CCSD(T) | High-Level Quantum Chemistry | Provides "gold-standard" reference energies for benchmarking and validation studies [9]. |
| CREST | Conformer Sampling Algorithm | Efficiently generates broad ensembles of molecular conformers using GFN-xTB methods [44] [4]. |
| GFN2-xTB | Semi-Empirical Quantum Method | Provides a fast and reasonably accurate potential energy surface for initial geometry sampling and optimization in CREST [4] [46]. |
| B97-3c | Composite Density Functional Theory | A cost-effective DFT method used for pre-screening and preliminary reoptimization of conformer ensembles [44] [45]. |
| ωB97X-D4 | Density Functional Theory | A robust, dispersion-corrected functional used for high-level optimization and final energy calculations [44] [45]. |
| MMFF94 | Molecular Mechanics Force Field | A high-accuracy force field for drug-like molecules, suitable for high-throughput conformational scoring [9]. |
| Reference Organic Molecule Set | Benchmark Dataset | A curated set of 145 molecules for validating conformational energy prediction methods [9]. |
This case study demonstrates that the choice of computational method profoundly impacts the accuracy of conformational energies for model drug fragments. While high-level QM methods like MP2 and DLPNO-CCSD(T) provide the highest accuracy, their computational expense often makes them impractical for routine use on large systems. The validation data shows that the B3LYP functional offers an excellent compromise, and for high-throughput scenarios, the MMFF94 and MM3-00 force fields provide the best performance among fast methods.
The development of efficient multi-level workflows, which use fast semi-empirical or force field methods for initial sampling and more accurate DFT methods for final refinement, represents the state of the art for handling drug-sized molecules [44] [4]. These workflows, rigorously validated against gold-standard reference data, provide a robust framework for achieving reliable conformational energy estimates, which are critical for successful structure-based drug design.
Overcoming Computational Challenges: Error Mitigation and Workflow Optimization
In the pursuit of accurate quantum chemical predictions for drug design and materials development, two systematic errors persistently challenge computational chemists: inadequate treatment of dispersion interactions and basis set superposition error (BSSE). Dispersion forces—weak attractive interactions arising from correlated electron fluctuations—are crucial for modeling van der Waals complexes, supramolecular assemblies, and biological systems, yet they remain poorly described by standard density functional theory (DFT) functionals [47]. Concurrently, BSSE emerges from the use of incomplete basis sets in supermolecular calculations, artificially enhancing binding energies when fragments borrow functions from interacting partners [48]. Within the broader thesis of validating conformational energies against post-Hartree-Fock methods, understanding and correcting these errors becomes paramount for achieving chemical accuracy (1 kcal/mol) in computational drug development workflows. This guide objectively compares contemporary correction strategies, providing researchers with validated protocols for reliable energetic predictions.
The Nature of Dispersion Interactions
Dispersion interactions, or van der Waals forces, originate from instantaneous multipole interactions between electrons in different molecular fragments. These critically important yet subtle forces significantly influence molecular conformation, crystal packing, host-guest interactions, and protein-ligand binding. Traditional local and semi-local DFT functionals fail to capture these correlations due to their inherent locality, systematically underestimating interaction energies in non-covalent complexes [47]. This deficiency manifests across multiple research domains, from overestimating interlayer distances in layered materials to inaccurate prediction of binding affinities in drug design.
The Basis Set Superposition Error (BSSE) Problem
BSSE arises fundamentally from the basis set incompleteness in quantum chemical calculations. In interaction energy computations using the supermolecular approach (ΔE = EAB - EA - EB), each monomer's energy calculation utilizes only its own basis functions, while the complex benefits from the combined, larger basis set. This imbalance allows monomers to artificially "borrow" functions from partners, lowering the complex's energy more than justified physically. The error becomes particularly pronounced with smaller basis sets lacking diffuse functions and significantly affects weak interactions where binding energies are comparable to BSSE magnitudes [48]. For neutral systems using triple-ζ basis sets, BSSE can still introduce notable inaccuracies, necessitating systematic correction for reliable results.
Methodological Approaches for Error Correction
Dispersion Correction Schemes
The computational chemistry community has developed multiple strategies to address DFT's dispersion deficiency:
Empirical Dispersion Corrections: Grimme's DFT-D methods add an empirical energy term (Edisp) based on atom-pairwise potentials with an R^−6 distance dependence. Popular implementations include D3 (with zero-damping or Becke-Johnson damping) and D4 versions, which incorporate some system-dependent polarizability considerations [49].
Van der Waals Density Functionals: Non-local functionals like VV10 incorporate dispersion directly into the exchange-correlation functional without empirical parameters, though at increased computational cost.
Hybrid Approaches: Composite methods like r²SCAN-3c integrate dispersion corrections with specialized basis sets for balanced performance [49].
Recent benchmarking studies on metal carbonyl complexes highlight how dispersion corrections significantly improve agreement with experimental structures and high-level reference data [49].
BSSE Correction and Basis Set Strategies
The counterpoise (CP) method, introduced by Boys and Bernardi, remains the most widely used BSSE correction approach [48]. It computes the BSSE for each monomer as the energy difference between calculations using its own basis set and the full complex basis set, providing a corrected interaction energy. However, CP correction increases computational cost and complexity, particularly for large systems.
Alternative Basis Set Strategies:
- Basis Set Extrapolation: Using exponential-square-root extrapolation schemes to approximate the complete basis set (CBS) limit can reduce BSSE dependence. Recent work optimized an extrapolation exponent (α = 5.674) for DFT calculations using def2-SVP and def2-TZVPP basis sets, achieving near-CBS accuracy with approximately half the computational time of CP-corrected calculations [48].
- Minimally-Augmented Basis Sets: Truhlar and coworkers developed "ma-" basis sets (e.g., ma-TZVP) with minimal diffuse functions that reduce BSSE while maintaining SCF convergence [48].
- Balanced Basis Set Selection: For neutral systems with triple-ζ basis sets and CP correction, studies indicate diffuse functions become unnecessary, simplifying computations [48].
Comparative Performance Analysis of Correction Methods
Benchmarking Dispersion-Corrected DFT Functionals
Table 1: Performance of Selected DFT Functionals with Dispersion Corrections for Structural and Energetic Properties
| Functional | Dispersion Correction | Performance for Geometries | Performance for Energetics | Recommended Use Cases |
|---|---|---|---|---|
| B3LYP | D3(BJ), D3(zero), D4 | Good | Moderate for interactions | General organic molecules |
| PBE | D3(BJ), D3(zero), D4 | Moderate | Moderate | Solid-state materials |
| PBE0 | D3(BJ), D3(zero), D4 | Good | Good | Thermochemistry |
| r²SCAN | D3(BJ), D4 | Excellent | Excellent | Balanced performance |
| TPSSh | D3(zero) | Excellent | Excellent | Organometallic complexes |
| ωB97X | D4 | Good | Good for excited states | Charge-transfer systems |
Recent comprehensive benchmarking of 54 functional/dispersion combinations for Mn(I) and Re(I) carbonyl complexes revealed that hybrid meta-GGA and meta-GGA functionals, particularly TPSSh(D3zero) and r²SCAN(D3BJ, D4), offer the optimal balance between accuracy and efficiency, providing reliable structures, vibrational properties, and energetics consistent with high-level DLPNO-CCSD(T) references [49].
BSSE Correction Efficiency Across Methods
Table 2: Efficiency of BSSE Mitigation Strategies for Weak Interaction Calculations
| Method | Basis Sets | BSSE Magnitude | Computational Cost | Accuracy vs. Reference |
|---|---|---|---|---|
| CP Correction | def2-TZVPP | Minimal | High | Excellent |
| CP Correction | def2-SVP | Significant | Low | Poor |
| Basis Set Extrapolation | def2-SVP/def2-TZVPP | Minimal | Moderate | Very Good |
| Minimal Augmentation | ma-TZVPP | Small | Moderate | Good |
| Standard Calculation | def2-QZVPP | Negligible | Very High | Excellent |
A systematic assessment of weak interaction energy calculations demonstrated that exponential-square-root extrapolation with optimized parameters achieves ~2% mean relative error compared to CP-corrected ma-TZVPP calculations while reducing computational time by approximately 50% [48]. This makes extrapolation particularly valuable for large-scale drug discovery applications.
Experimental Protocols for Method Validation
Protocol 1: Benchmarking Dispersion Corrections for Conformational Energies
Objective: Validate dispersion-corrected DFT methods against high-level reference data for organic molecule conformational energies.
Reference Standard: DLPNO-CCSD(T)/cc-pVTZ calculations provide reference conformational energies [40]. A benchmark set of 145 organic molecules with 158 conformational energies and barriers represents typical fragments in drug-like molecules [40].
Procedure:
- Select diverse organic molecules representing drug fragments (hydrocarbons, haloalkanes, O/N/P/S-containing compounds)
- Generate low-energy conformers using conformational sampling algorithms
- Optimize geometries at DFT level with various dispersion corrections
- Calculate single-point energies at DLPNO-CCSD(T)/cc-pVTZ level for benchmark
- Compute mean absolute errors (MAE) and root-mean-square errors (RMSE) for each method
Expected Outcomes: Recent benchmarking shows MP2 achieves lowest error (MAE = 0.35 kcal/mol), followed by B3LYP (0.69 kcal/mol), and HF theories (0.81-1.0 kcal/mol). Among force fields, MM3-00 (1.28 kcal/mol) and MMFF94 (1.30 kcal/mol) perform best, while universal force fields like UFF show significantly higher errors (3.77 kcal/mol) [40].
Protocol 2: Assessing BSSE Correction Accuracy
Objective: Quantify BSSE effects and correction efficacy for intermolecular complexes.
Reference Systems: Utilize the S66, S30L, and related benchmark sets containing diverse non-covalent interactions.
Procedure:
- Extract complex geometries from benchmark datasets
- Compute interaction energies at target level (e.g., B3LYP-D3(BJ)/def2-TZVPP)
- Calculate CP-corrected interaction energies using the standard protocol
- Compute basis set extrapolated energies using def2-SVP/def2-TZVPP with α = 5.674
- Compare with reference CCSD(T)/CBS values
- Statistical analysis: calculate MAE, RMSE, and maximum deviations
Validation: The optimized extrapolation parameter (α = 5.674) for B3LYP-D3(BJ) provides near-identical results to CP-corrected ma-TZVPP calculations with significantly reduced computational cost [48].
Figure 1: BSSE Correction Methodology Workflow. This diagram illustrates the comparative workflow for evaluating basis set superposition error correction methods against high-level quantum chemical references.
Table 3: Key Research Reagent Solutions for Error-Corrected Quantum Calculations
| Tool/Resource | Type | Function | Implementation Examples |
|---|---|---|---|
| Grimme's D3/D4 | Dispersion Correction | Adds empirical dispersion correction to DFT functionals | ORCA, Gaussian, Q-Chem, Sherpa |
| Counterpoise Method | BSSE Correction | Corrects for basis set superposition error in interaction energies | Built-in in most major quantum codes |
| Basis Set Extrapolation | BSSE Mitigation | Approximates complete basis set limit using smaller basis sets | Custom scripts, ORCA automation |
| DLPNO-CCSD(T) | Reference Method | Provides gold-standard reference energies for method validation | ORCA, Molpro |
| Benchmark Sets | Validation Data | Standardized molecular sets for method comparison (S66, S30L, etc.) | Publicly available datasets |
| r²SCAN Functional | Density Functional | Meta-GGA functional with excellent performance across diverse systems | ORCA, VASP, Quantum ESPRESSO |
Integration in Drug Discovery Workflows
Accurate conformational energy prediction directly impacts virtual screening, binding affinity prediction, and polymorph stability assessment in pharmaceutical development. Machine learning approaches now offer promising alternatives, with deep learning-based density functional methods (DeePHF, DeePKS) achieving chemical accuracy for molecular energies of drug-like molecules with limited CCSD(T)/def2-TZVP training data [50]. These methods demonstrate excellent transferability, potentially bypassing traditional systematic error corrections while maintaining high precision.
The emerging paradigm combines robust error correction for specific challenging cases with machine learning acceleration for high-throughput screening. For drug development professionals, this translates to more reliable prediction of protein-ligand binding modes, more accurate assessment of relative conformational stability, and improved virtual screening outcomes through reduced systematic biases in energy calculations.
Systematic errors from inadequate dispersion treatment and basis set superposition remain significant challenges in computational chemistry, but rigorous correction strategies now enable DFT-level calculations to achieve near-chemical accuracy for conformational energies and interaction energies. Dispersion-corrected hybrid meta-GGA functionals like TPSSh and r²SCAN combined with optimized BSSE mitigation through basis set extrapolation provide an excellent balance of accuracy and computational efficiency for drug discovery applications.
Future methodological developments will likely focus on integrated machine learning potentials trained on high-level reference data, potentially obviating the need for explicit a posteriori error corrections. Meanwhile, current best practices recommend careful functional validation against DLPNO-CCSD(T) references for specific chemical systems of interest, ensuring reliable predictions in pharmaceutical development pipelines.
In computational chemistry, the trade-off between the computational cost of a method and its accuracy is a fundamental consideration, especially for large-scale or high-throughput studies. This balance is critically important in fields like drug development, where reliable predictions of molecular properties can significantly accelerate discovery pipelines. The core challenge lies in the scaling behavior of quantum chemistry methods; while post-Hartree-Fock (post-HF) methods like CCSD(T) are considered the "gold standard" for accuracy, their prohibitive computational cost (scaling as O(N⁷) with system size) makes them impractical for screening large molecular libraries [51]. Conversely, more approximate methods like Density Functional Theory (DFT) offer faster computation but can suffer from systematic errors in key chemical regimes, such as long-range charge transfer, non-covalent interactions, and transition-metal complexes [51]. This guide objectively compares the performance of different computational strategies, focusing on their application for validating conformational energies within a high-throughput framework.
Performance Comparison of Computational Methods
Quantitative Comparison of Method Accuracy and Cost
The following table summarizes the key performance characteristics of prevalent computational methods, highlighting the inherent trade-off between computational expense and accuracy.
Table 1: Performance Comparison of Quantum Chemistry Methods for Molecular Property Prediction
| Method | Typical Scaling | Computational Cost | Key Strengths | Key Limitations | Suitable for High-Throughput? |
|---|---|---|---|---|---|
| Coupled Cluster (e.g., CCSD(T)) | O(N⁷) | Prohibitively high for molecules >32 atoms [51] | Gold-standard accuracy for thermochemistry, kinetics [51] | Astronomical cost for peptides/drug complexes [51] | No, cost is prohibitive |
| Density Functional Theory (DFT) | O(N³) to O(N⁴) | Moderate to high for large systems | Reasonable cost/accuracy balance; widely used [52] | Systematic errors in charge transfer, non-covalent interactions, transition metals [51] | Limited, requires careful functional selection |
| Neural Network Wavefunctions (LWMs) | Varies (inference cost is critical) | Lower cost for data generation after training [51] | Approaches exact solution; handles static/dynamic correlation [51] | Training complexity; emerging technology | Yes, enables affordable gold-standard datasets [51] |
| Machine Learning Interatomic Potentials (MLIPs) | ~O(N) (classical simulation speeds) | Very low after training [51] | Ab-initio accuracy at classical speeds [51] | Dependent on quality and scope of training data | Yes, for ultra-fast screening on large scales |
Emerging Solutions and Benchmarking Data
Recent algorithm-hardware co-designs and novel computational paradigms demonstrate significant advances in mitigating the cost-accuracy trade-off.
Table 2: Emerging Strategies for Enhancing Efficiency and Accuracy
| Strategy/Technology | Reported Efficiency Gain | Reported Accuracy Impact | Key Innovation |
|---|---|---|---|
| T8HWQ for SNN on FPGA [53] | 6x throughput improvement vs. other SNN accelerators [53] | <0.7% accuracy degradation on CIFAR-100 vs. full-precision [53] | Algorithm-hardware co-design unifying compute patterns. |
| simulacra AI's LWM Pipeline [51] | 15-50x cost reduction for data gen. vs. Microsoft pipeline; 2-3x vs. traditional CCSD on amino acids [51] | Parity in energy accuracy with state-of-the-art pipelines [51] | Large Wavefunction Models with novel RELAX sampling. |
| Physics-Informed ML for Materials [52] | Outperforms state-of-the-art in prediction/optimization efficiency [52] | Improved prediction accuracy with physical interpretability [52] | Integrates physical principles into deep learning. |
Experimental Protocols for Method Validation
Foundational Principles of Benchmarking
A rigorous method comparison study must be carefully designed to yield unbiased and informative results. The following guidelines are essential [54]:
- Define Purpose and Scope: Clearly state whether the study is a neutral comparison or for demonstrating a new method's merits. Neutral benchmarks should be as comprehensive as possible [54].
- Selection of Methods: Include all relevant, available methods. Justify the exclusion of any widely used methods. For new method papers, compare against current best-performing and baseline methods [54].
- Selection of Datasets: Use benchmark datasets that cover the entire clinically or chemically meaningful range. A combination of simulated data (with known ground truth) and real experimental data is often ideal. A minimum of 40, and preferably 100, samples is recommended for robust comparison [54] [55].
- Avoid Inadequate Statistical Tests: Correlation analysis (e.g., Pearson's r) only measures linear association, not agreement, and can be misleading. T-tests are also inadequate for assessing comparability, as they may miss clinically meaningful differences or find statistically significant but trivial ones [55].
Workflow for Computational Method Validation
The diagram below outlines a generalized experimental workflow for validating and comparing computational chemistry methods, incorporating best practices from benchmarking studies.
Diagram 1: Workflow for validating computational methods, integrating benchmarking principles and performance analysis.
Detailed Protocol: Validating a New Low-Cost Method
For a study aiming to validate a new, cost-efficient method (e.g., a Machine Learning Potential or a Large Wavefunction Model) against high-accuracy benchmarks, the following detailed protocol is recommended:
Reference Method and Dataset Curation:
- Select a high-accuracy reference method, typically CCSD(T) with a complete basis set (CBS) limit, where feasible for the system size [51].
- Curate a diverse dataset of molecular conformations. The dataset must cover a wide energy range and include chemically relevant non-covalent interactions, torsional strains, and transition states if applicable.
- Compute reference conformational energies for this dataset using the high-accuracy method. This becomes the ground truth for the benchmark.
Execution of Candidate Methods:
- Run the same set of conformational energy calculations using the candidate methods (e.g., various DFT functionals, the new ML potential, or LWM).
- For DFT, use standardized parameters (functional, basis set, convergence criteria) across all calculations. Do not perform extensive parameter tuning for some methods while using defaults for others, as this introduces bias [54].
- For ML-based methods, ensure the benchmark molecules are not part of the training set to properly assess generalizability.
Performance Analysis and Metrics:
- Primary Metric - Energy Accuracy: Calculate the mean absolute error (MAE) and root mean square error (RMSE) of the predicted conformational energies relative to the reference method for each candidate.
- Primary Metric - Statistical Efficiency: For stochastic methods (e.g., Variational Monte Carlo), report the effective sample size (ESS) and autocorrelation time, as these critically impact the true computational cost and reliability of results [51].
- Secondary Metric - Computational Cost: Measure the total computational time, core-hours, or memory footprint for each method. Correlate this with the achieved accuracy.
- Data Visualization: Create scatter plots of predicted vs. reference energies to identify systematic errors and Bland-Altman-like difference plots (or direct difference vs. reference energy plots) to visualize bias across the energy range [55].
The Scientist's Toolkit: Essential Research Reagents and Solutions
This section details key computational "reagents" and tools essential for conducting high-throughput computational studies.
Table 3: Essential Computational Tools for High-Throughput Studies
| Tool / Solution | Function / Description | Application Context |
|---|---|---|
| Density Functional Theory (DFT) | A computational method for electronic structure calculation using functionals of the electron density. | Provides a baseline for energy and property prediction; balance of cost and accuracy [56] [52]. |
| Coupled Cluster Theory (CCSD(T)) | A post-Hartree-Fock method considered the "gold standard" for single-reference quantum chemistry. | Provides benchmark-quality reference data for training and validation [51]. |
| Large Wavefunction Models (LWMs) | Foundation neural-network wavefunctions optimized by Variational Monte Carlo (VMC). | Generates near-exact quantum data at a fraction of the cost of traditional CCSD(T) [51]. |
| Machine Learning Interatomic Potentials (MLIPs) | Machine-learned potentials that mimic ab-initio accuracy at classical force-field speeds. | Enables ultra-fast molecular dynamics and property screening for very large systems [51]. |
| High-Throughput Computing (HTC) Workflows | Automated computational pipelines for managing large-scale simulations on distributed systems. | Essential for screening vast material libraries and generating massive datasets [52]. |
| Physics-Informed Machine Learning | ML models that incorporate physical laws or constraints directly into the learning process. | Improves prediction accuracy, generalizability, and interpretability of data-driven models [52]. |
| Statistical Analysis Software (R/Python) | Environments for implementing statistical analyses, data visualization, and regression models. | Critical for performing rigorous method comparison and quantifying uncertainty [57]. |
The landscape of computational strategies for large-scale studies is evolving beyond the simple dichotomy of "accurate but expensive" versus "fast but approximate." Traditional benchmarks clearly establish the hierarchy from DFT to post-HF methods, with CCSD(T) remaining the reference for accuracy. However, emerging paradigms like Large Wavefunction Models and specialized algorithm-hardware co-designs are fundamentally altering the cost-accuracy curve. These approaches demonstrate that it is possible to achieve gold-standard accuracy at a dramatically reduced computational cost, making high-throughput, high-fidelity screening of molecular systems a tangible reality. For researchers in drug development, the strategic integration of these tools—using fast methods for initial screening and high-accuracy methods for final validation, or leveraging LWMs to generate affordable training data for MLIPs—offers a robust path to accelerating discovery while maintaining confidence in computational predictions.
In contemporary computational chemistry, particularly within the domain of density functional theory (DFT), the arbitrary selection of theoretical methods no longer meets the standards required for publication in high-impact journals. The performance of density functional approximations (DFAs) exhibits significant variability across different chemical systems and properties, making method justification an essential component of rigorous scientific reporting. As highlighted by recent benchmarking efforts, functional accuracy displays profound context dependence, exemplified by the fact that top-performing functionals for hydrogen-bonding interactions differ markedly from those optimal for general conformational analysis [58]. This methodological review establishes a structured framework for researchers to justify computational protocols, particularly focusing on the validation of conformational energies against post-Hartree-Fock (post-HF) reference methods, thereby addressing a critical need for standardized reporting practices in computational drug development and materials science.
The evolution of DFT has produced a complex ecosystem of functional approximations, often described as a "Charlotte's Web" of interconnected methodologies [59]. This diversity, while enabling specialized applications, simultaneously necessitates rigorous validation protocols to ensure methodological appropriateness for specific research questions. Within pharmaceutical development, where computational predictions increasingly guide experimental resource allocation, the justification of DFT methodologies against higher-level wavefunction theories has transitioned from optional to mandatory practice. This guide provides both theoretical framework and practical protocols for establishing this methodological justification, with particular emphasis on conformational energy predictions—a property critically important to molecular recognition, binding affinity, and ultimately, therapeutic efficacy.
Comparative Performance Analysis of Computational Methods
Density Functional Performance Across Chemical Systems
Table 1: Performance of Select Density Functional Approximations Across Benchmark Systems
| Functional | Functional Type | Hydrogen Bonding MAE (kcal/mol) [58] | Conformational Sampling Efficiency | Recommended Application Domains |
|---|---|---|---|---|
| B97M-V | meta-GGA | < 0.5 (Top Performer) | Moderate | Non-covalent interactions, supramolecular assembly |
| B97M-D3(BJ) | meta-GGA+Dispersion | ~0.5 (Top Performer) | Moderate | Multiple hydrogen-bonded systems, self-assembly |
| ωB97X-V | Range-Separated Hybrid | Not specified | Low (High cost) | Charge-transfer systems, excited states [4] |
| B3LYP | Global Hybrid | > 1.0 (Variable performance) | Moderate | General purpose, organic molecules |
| B97-3c | Composite DFT | Not specified | High (Low cost) | Conformational ensemble pre-optimization [4] |
| GFN2-xTB | Semi-empirical | Not specified | Very High | Initial conformational searching, large systems [46] |
The benchmarking data reveals several critical trends for computational researchers. For non-covalent interactions, particularly the challenging case of quadruple hydrogen bonds, the Berkeley family of functionals (B97M-V and variants) consistently delivers superior performance, with mean absolute errors (MAE) below 0.5 kcal/mol compared to CCSD(T)-cf reference data [58]. This exceptional accuracy is attributed to their sophisticated treatment of medium-range correlation effects and incorporation of appropriate dispersion corrections. For conformational sampling tasks, efficiency considerations become paramount, with composite methods (e.g., B97-3c) and semi-empirical approaches (GFN2-xTB) offering viable strategies for initial exploration before refinement with more accurate functionals [4] [46].
The critical importance of system-specific functional selection is further exemplified by the performance variations observed across different interaction types. Functionals like B3LYP, while historically popular for general organic applications, demonstrate variable performance for non-covalent interactions, particularly without appropriate dispersion corrections [59] [58]. Range-separated hybrids such as ωB97X-V offer superior performance for charge-transfer systems and excited states but at substantially increased computational cost [4]. These observations underscore the fundamental principle that functional selection must be guided by both the specific chemical system under investigation and the target molecular properties, rather than historical precedent or computational convenience alone.
Multi-level Method Comparison and Benchmarking Strategies
Table 2: Hierarchy of Computational Methods for Validation Studies
| Method Class | Representative Methods | Typical Application | Relative Cost | Key Limitations |
|---|---|---|---|---|
| Coupled Cluster | DLPNO-CCSD(T)-cf [58] | Gold-standard reference | Extremely High | System size limitations, computational expense |
| Double Hybrid DFT | DSD-BLYP, ωB97X-2LYP | High-accuracy validation | High | Limited availability in codes, system size limits |
| Hybrid DFT | B97M-V, ωB97X-D4 | Production calculations | Moderate-High | Cost scaling with system size |
| Meta-GGA DFT | B97M-V, SCAN, TPSS | Balanced accuracy/cost | Moderate | Self-interaction error |
| GGA DFT | BLYP, PBE | Initial screening | Low-Moderate | Poor dispersion treatment |
| Semi-empirical | GFN2-xTB, GFN-FF | Large system sampling | Very Low | Parameter transferability |
Modern validation protocols require careful navigation of this methodological hierarchy. The continued-fraction coupled-cluster [CCSD(T)-cf] approach represents the current gold standard for reference data, particularly for non-covalent interactions where high-level electron correlation treatment is essential [58]. For larger systems where coupled cluster calculations become prohibitive, double-hybrid functionals and domain-based local approximations (DLPNO) provide viable alternatives with favorable accuracy-to-cost ratios. The critical protocol justification emerges from systematically demonstrating that production methods (typically hybrid or meta-GGA DFT) reproduce trends and absolute values established by these higher-level references within acceptable error margins—typically chemical accuracy (1 kcal/mol) for energy differences.
The benchmark study on quadruple hydrogen bonds exemplifies this approach, testing 152 density functional approximations against CCSD(T)-cf reference values to establish reliable performers for specific interaction types [58]. Similarly, workflow validation for conformational sampling establishes reliability through comparison against established benchmarks and experimental data where available [4] [60]. This multi-level strategy ensures that methodological choices are not arbitrary but are instead grounded in empirical performance data relevant to the specific chemical context under investigation.
Experimental Protocols for Method Validation
Benchmarking Workflow for Functional Validation
Diagram 1: Functional Benchmarking Workflow. This protocol establishes a systematic approach for identifying optimal density functional approximations (DFAs) for specific applications through comparison with high-accuracy reference data.
The benchmarking workflow initiates with careful definition of the benchmark set, which should represent the key chemical motifs relevant to the target application. For drug-like molecules, this typically includes diverse non-covalent interactions, conformational flexibilities, and electronic properties characteristic of pharmaceutical compounds. The subsequent reference calculation phase employs high-level wavefunction methods (e.g., DLPNO-CCSD(T) extrapolated to complete basis set limit) to establish reliable reference data [58]. The critical benchmarking phase evaluates multiple density functional approximations against these references, with particular attention to statistical measures like mean absolute error (MAE) and identification of systematic outliers. This rigorous approach transforms functional selection from an arbitrary choice to an empirically justified decision, meeting modern publication standards for computational methodology.
Conformational Ensemble Generation Protocol
Diagram 2: Conformational Ensemble Generation. This multi-level workflow efficiently generates reliable conformational ensembles through sequential refinement, balancing computational cost with quantum chemical accuracy.
The conformational ensemble protocol addresses a fundamental challenge in computational drug discovery: efficiently exploring conformational space while maintaining quantum chemical accuracy. The workflow begins with extensive initial conformer generation using specialized sampling tools like CREST, which employs metadynamics with the GFN2-xTB semi-empirical method to ensure comprehensive coverage of conformational space [4]. The critical pre-optimization and deduplication stage eliminates spurious minima and reduces redundant calculations, with composite methods like B97-3c offering favorable cost-accuracy balance for this purpose [4]. Subsequent DFT refinement with appropriately selected functionals (e.g., ωB97X-D4) provides quantum mechanical accuracy, while final single-point calculations with larger basis sets (def2-QZVPP) yield precise energies for Boltzmann weighting and property prediction. This protocol represents current best practice for conformational analysis of flexible drug-like molecules.
Table 3: Research Reagent Solutions for Computational Validation
| Tool Category | Specific Tools | Primary Function | Application in Validation |
|---|---|---|---|
| Quantum Chemistry Software | Psi4 [58], ORCA [4] | Electronic structure calculations | Reference and production calculations |
| Conformational Sampling | CREST [4], GOAT [4] | Conformer ensemble generation | Comprehensive conformational space exploration |
| Semi-empirical Methods | GFN2-xTB [46], GFN-FF [46] | Rapid geometry optimization | Initial screening and large system treatment |
| Composite Methods | B97-3c [4], r2SCAN-3c [4] | Balanced accuracy/cost calculations | Intermediate optimization steps |
| Benchmark Databases | S22, S66, S66x8 [58], PubChemQCR [61] | Reference data sources | Method validation and benchmarking |
| Plane-Wave DFT Codes | ADF [62] | Periodic boundary calculations | Solid-state and material system studies |
| Machine Learning Potentials | DeePHF, DeePKS [50] | Accelerated quantum calculations | High-throughput screening applications |
The modern computational chemist's toolkit encompasses diverse software solutions and theoretical methods, each with specific strengths for validation protocols. Specialized conformational sampling tools like CREST implement advanced algorithms (RMSD-biased metadynamics) to overcome energy barriers and ensure comprehensive phase space coverage [4]. Quantum chemistry packages such as Psi4 and ORCA provide implementations of high-level wavefunction methods and density functional approximations necessary for reference calculations and production work [58] [4]. Emerging machine learning potentials (e.g., DeePHF, DeePKS) represent promising approaches for combining DFT-level accuracy with significantly reduced computational cost, particularly valuable for high-throughput screening applications [50].
The critical importance of benchmark databases cannot be overstated, as these curated collections provide essential reference data for method validation. Established datasets like S22, S66, and S66x8 provide non-covalent interaction benchmarks [58], while emerging resources like PubChemQCR offer extensive relaxation trajectories for organic molecules [61]. These repositories enable researchers to establish method performance before application to novel systems, fulfilling the fundamental requirement of demonstrated methodological appropriateness for specific chemical problems—a cornerstone of modern computational reporting standards.
This guide has established a comprehensive framework for justifying computational protocols in published research, with specific application to conformational energy validation using DFT and post-HF methods. The presented data unequivocally demonstrates that functional performance is system-dependent, necessitating empirical validation rather than assumed transferability. The benchmark results indicate that specialized functionals like B97M-V deliver superior performance for specific interactions like hydrogen bonding, while multi-level workflows combining semi-empirical methods with DFT refinement provide optimal balance for conformational sampling tasks.
The experimental protocols and benchmarking strategies detailed herein provide actionable approaches for establishing methodological rigor, satisfying the increasing demands of journal reviewers and editors for justified computational approaches. As computational chemistry continues to expand its role in pharmaceutical development and materials design, the adoption of these validation-centered practices will ensure published results meet the highest standards of reliability and reproducibility. By implementing these protocols, researchers can confidently select and justify computational methods, advancing both their immediate research objectives and the broader standards of computational molecular science.
Computational chemistry provides a hierarchy of methods for modeling molecular systems, each balancing accuracy and computational cost. Force fields, density functional theory (DFT), and high-level wavefunction theory (WFT) represent different rungs on this ladder. While each can be used independently, multi-level approaches that strategically combine these methods offer a powerful paradigm for tackling complex problems efficiently, particularly for challenging properties like conformational energies in transition metal complexes and drug-like molecules. These hybrid protocols leverage the speed of lower-level methods with the precision of high-level computations, creating workflows that are both computationally tractable and chemically accurate. This guide examines the performance, protocols, and practical applications of these multi-level strategies, providing researchers with a framework for their implementation in computational chemistry and drug discovery.
Performance Benchmarking of Computational Methods
Accuracy of Methods for Transition Metal Complexes
Transition metal complexes present particular challenges due to their complex electronic structures. Benchmark studies against curated experimental data reveal significant performance differences between methods.
Table 1: Performance of Quantum Chemistry Methods for Transition Metal Spin-State Energetics (SSE17 Benchmark)
| Method Category | Specific Methods | Mean Absolute Error (kcal mol⁻¹) | Maximum Error (kcal mol⁻¹) | References |
|---|---|---|---|---|
| Coupled Cluster | CCSD(T) | 1.5 | -3.5 | [63] |
| Double-Hybrid DFT | PWPB95-D3(BJ), B2PLYP-D3(BJ) | <3.0 | <6.0 | [63] |
| Multireference WFT | CASPT2, MRCI+Q, CASPT2/CC, CASPT2+δMRCI | Variable, outperformed by CCSD(T) | Variable, outperformed by CCSD(T) | [63] |
| Popular DFT for Spin States | B3LYP*-D3(BJ), TPSSh-D3(BJ) | 5-7 | >10 | [63] |
For conformational energies of transition metal complexes (TMCONF40 benchmark), double-hybrid DFT references with low-cost composite methods show strong performance, with Pearson correlation coefficients of rₚ,mean,B97-3c//B97-3c = 0.922 and rₚ,mean,PBEh-3c//B97-3c = 0.890, and mean absolute deviations close to or below 1 kcal mol⁻¹ [64]. The GFN2-xTB semi-empirical method outperforms PMx methods but shows more moderate correlations (rₚ,mean,GFN2-xTB//B97-3c = 0.567, MADmean = 2.68 kcal mol⁻¹) [64].
Performance Across Organic Molecules and Drug-like Compounds
For organic molecules, machine learning potentials like ANI-1ccx and ANI-2x demonstrate remarkable accuracy in predicting torsional energy profiles, effectively capturing minimum and maximum values of these profiles [65]. These neural network methods, trained on coupled-cluster and DFT data respectively, offer a compelling alternative to traditional quantum chemistry methods for conformational analysis.
Table 2: Method Performance for Conformational Energies of Organic Molecules
| Method | Theoretical Foundation | Computational Speed | Key Strengths | Limitations |
|---|---|---|---|---|
| ANI-1ccx | CCSD(T)/CBS-trained ML | Very Fast | High accuracy for torsional profiles | Limited to H, C, N, O elements |
| ANI-2x | ωB97X/6-31G(d)-trained ML | Very Fast | Good accuracy; includes H, C, N, O, F, Cl, S | Limited element set |
| Double-Hybrid DFT | DFT with MP2 correlation | Moderate | High accuracy for diverse systems | Computationally demanding |
| Hybrid DFT (B3LYP) | DFT with HF exchange | Moderate | Balanced cost/accuracy | Weak on van der Waals interactions |
| GFN2-xTB | Semi-empirical tight binding | Fast | Good for initial screening | Lower accuracy for complex interactions |
Traditional DFT methods with the B3LYP functional and OPLS force fields often show deviations from more accurate methods in conformational energy predictions, as they "weakly consider van der Waals and other intramolecular forces in torsional energy profiles" [65]. The ANI models account for non-bonded intramolecular interactions more effectively, making them particularly suitable for drug discovery applications where conformational flexibility impacts binding.
Experimental Protocols and Workflows
Multi-Level Conformational Sampling Protocol
The CREST (Conformer-Rotamer Ensemble Sampling Tool) protocol implements a multi-level approach for generating conformer ensembles of transition metal complexes [64]:
Figure 1: CREST multi-level conformational sampling workflow for transition metal complexes.
This workflow begins with meta-dynamics (MTD) sampling using GFN2-xTB, which broadly explores the potential energy surface. The sampled structures then undergo geometry optimization at the same GFN2-xTB level. Subsequent energetic pre-screening using GFN2-xTB single-point energies identifies promising candidates, which are finally re-ranked with high-level methods such as DLPNO-CCSD(T1) or double-hybrid DFT [64]. This protocol demonstrates how semi-empirical methods can efficiently handle the computationally demanding sampling step, while more accurate—but expensive—methods are reserved for the final energetic ranking of a reduced set of candidates.
Hybrid DFT//GFN2-xTB Force Field Parameterization
For molecular dynamics simulations, accurate force field parameters are essential. A hybrid quantum mechanical workflow has been developed for custom torsion parameterization [66]:
Figure 2: Hybrid workflow for force field parameterization.
This protocol begins with Wiberg Bond Order (WBO)-based fragmentation, which preserves the electronic environment around rotatable bonds while reducing system size. The workflow performs torsional scans at GFN2-xTB level (15° intervals) followed by B3LYP-D3BJ/DZVP single-point energy calculations on each rotamer geometry [66]. Validation across 550+ small molecules shows this approach reproduces full quantum mechanical torsion scans within 1.08 kcal/mol while achieving a 4-12× computational speedup compared to full QM calculations [66]. The resulting parameters significantly improve conformational sampling in molecular dynamics (MD) and free energy perturbation (FEP) simulations.
QTAIM-Enhanced Machine Learning Workflow
Integrating quantum chemical descriptors with machine learning provides another powerful multi-level approach. The QTAIM-enriched graph neural network workflow combines quantum theory of atoms in molecules with machine learning [67]:
- Dataset Preparation: The tmQM+ dataset of 60k transition metal complexes provides structures with varied charges, spins, and metal centers
- QTAIM Descriptor Calculation: DFT calculations followed by QTAIM analysis yield electronic structure descriptors
- Graph Neural Network Training: Molecular graphs are constructed using QTAIM bond critical points, featurized with quantum descriptors
- Property Prediction: The trained model predicts molecular properties with DFT-level accuracy at significantly reduced computational cost
This approach demonstrates that "additional quantum chemical information improves performance on unseen regimes and smaller training sets" [67], enabling accurate predictions for complexes beyond the training data distribution.
Research Reagent Solutions
Successful implementation of multi-level computational approaches requires familiarity with specialized software tools and computational resources.
Table 3: Essential Research Reagents for Multi-Level Computational Chemistry
| Tool Name | Type | Primary Function | Application Context |
|---|---|---|---|
| CREST | Software Package | Conformer sampling with GFNn-xTB | Automated conformer ensemble generation for transition metal complexes [64] |
| GFN2-xTB | Semi-empirical Method | Fast geometry optimization and energy calculation | Initial sampling, pre-screening, and molecular dynamics [64] [66] |
| ORCA | Quantum Chemistry | DFT, coupled cluster, and DLPNO-CCSD(T) calculations | High-level single-point energies and reference calculations [63] [67] |
| Q-Chem | Quantum Chemistry | DFT with comprehensive functional library | Exchange-correlation functional development and application [68] |
| Multiwfn | Analysis Tool | QTAIM and electronic structure analysis | Quantum descriptor generation for machine learning [67] |
| qtaim-embed | ML Package | Graph neural networks for molecules | Property prediction with QTAIM-enriched features [67] |
| ANI-1ccx/ANI-2x | ML Potentials | Neural network potential energy surfaces | Fast conformational energy predictions for organic molecules [65] |
| TMCONF40 | Benchmark Set | 40 transition metal complex conformers | Method validation and development [64] |
| SSE17 | Benchmark Set | 17 transition metal spin-state energetics | Experimental validation of quantum methods [63] |
These "research reagents" form the foundation for implementing the multi-level approaches described in this guide. The benchmark sets (TMCONF40, SSE17) play a particularly crucial role in method validation, providing curated data for assessing computational protocols against reliable reference values [63] [64].
Multi-level computational approaches represent a sophisticated strategy for balancing accuracy and efficiency in computational chemistry. By leveraging the respective strengths of force fields, semi-empirical methods, DFT, and high-level wavefunction theory, researchers can tackle complex problems ranging from transition metal conformational energetics to drug discovery. The protocols and benchmarks presented here provide a roadmap for implementing these strategies effectively, with validation data ensuring methodological reliability. As computational demands grow with system complexity and the need for higher accuracy, these hierarchical approaches will become increasingly essential tools in the computational chemist's toolkit.
Benchmarking Performance: A Systematic Validation of Methods Against High-Level References
The accurate prediction of conformational energies is a cornerstone of computational chemistry, with profound implications for drug design, materials science, and molecular dynamics simulations. The reliability of these predictions hinges on the computational methods employed, each offering distinct trade-offs between accuracy and computational cost. This review provides a quantitative performance assessment of prevalent quantum mechanical (QM) methods—including Density Functional Theory (DFT) and post-Hartree-Fock (post-HF) approaches—alongside classical molecular mechanics force fields, based on their mean errors in conformational energy predictions. Framed within the broader thesis of validating computational methodologies, this analysis leverages benchmarking against high-level coupled cluster theory to offer researchers a clear, data-driven guide for method selection in conformational energy studies, particularly for drug-like molecules.
The table below synthesizes key quantitative findings from a comprehensive benchmarking study that evaluated common computational methods against the DLPNO-CCSD(T)/cc-pVTZ level of theory, which served as the reference for 158 conformational energies of 145 organic molecules relevant to drug design [40].
Table 1: Mean Absolute Errors (MAE) in Conformational Energy Predictions
| Method Category | Specific Method | Mean Absolute Error (kcal mol⁻¹) | Computational Cost |
|---|---|---|---|
| Post-Hartree-Fock | MP2 | 0.35 | Very High |
| CCSD(T) | (Reference) | Extremely High | |
| Density Functional Theory | B3LYP | 0.69 | Medium |
| Hartree-Fock | HF (various basis sets) | 0.81 - 1.00 | Low-Medium |
| Advanced Force Fields | MMFF94 | 1.30 | Very Low |
| MM3-00 | 1.28 | Very Low | |
| MM3-96 | 1.40 | Very Low | |
| Traditional Force Fields | MMX | 1.77 | Very Low |
| MM+ | 2.01 | Very Low | |
| MM4 | 2.05 | Very Low | |
| Generic Force Fields | DREIDING | 3.63 | Very Low |
| UFF | 3.77 | Very Low |
The data reveals a clear performance-accuracy trade-off. Post-HF methods, particularly MP2, demonstrate superior accuracy with a mean error of just 0.35 kcal mol⁻¹, establishing it as a high-accuracy benchmark, though its computational expense limits application to large systems [40]. DFT, represented by the popular B3LYP functional, offers a practical balance with respectable accuracy (0.69 kcal mol⁻¹) and significantly lower computational cost than MP2 [40]. Among force fields, a significant performance spread exists based on their parameterization; advanced force fields like MMFF94 and MM3-00 achieve errors near 1.3 kcal mol⁻¹, making them suitable for high-throughput screening of drug-sized molecules, whereas more generic force fields like UFF and DREIDING show substantially larger errors (>3.6 kcal mol⁻¹) [40].
Experimental Protocols for Benchmarking
The quantitative data presented in this review is derived from rigorously designed benchmarking studies. Understanding their protocols is essential for contextualizing the results.
Molecular Data Set and Reference Values
The foundational benchmark study selected 145 reference organic molecules comprising model fragments commonly encountered in computer-aided drug design [40]. This set included hydrocarbons, haloalkanes, conjugated compounds, and oxygen-, nitrogen-, phosphorus-, and sulphur-containing compounds, ensuring broad chemical diversity [40]. The study calculated 158 conformational energies and barriers. The key to this benchmark's reliability is its use of Domain-based Local Pair Natural Orbital Coupled Cluster (DLPNO-CCSD(T)) theory with cc-pVTZ basis sets as the source for reference conformational energies [40]. DLPNO-CCSD(T) is considered a "gold standard" for its high accuracy while being computationally more feasible than canonical CCSD(T) for larger molecules, thus providing a credible reference for evaluating more efficient methods [40].
Benchmarking Procedure
The tested methods were evaluated by their ability to reproduce the DLPNO-CCSD(T) reference conformational energies. The core protocol can be summarized as follows [40]:
- Conformer Selection: Low-energy conformers and transition states for the 145 organic molecules were identified.
- Reference Calculation: Single-point energy calculations at the DLPNO-CCSD(T)/cc-pVTZ level were performed on these structures to establish the reference energies.
- Method Evaluation: For each method (force fields, HF, DFT), the conformational energies were computed.
- Error Analysis: The mean absolute error (MAE) for each method was calculated by comparing its predicted conformational energies against the DLPNO-CCSD(T) reference values.
This workflow ensures a consistent and fair comparison across different methodological paradigms, from quantum mechanics to molecular mechanics.
Figure 1: Workflow for benchmarking computational methods against a high-level quantum chemistry reference. The process begins with selecting a diverse set of molecules and calculating reference energies using a gold-standard method like DLPNO-CCSD(T). Target methods are then evaluated by computing their mean absolute error (MAE) against this reference.
Quantum Chemical Methods
The high accuracy of MP2 (MAE: 0.35 kcal mol⁻¹) stems from its explicit, though perturbative, treatment of electron correlation, which is crucial for capturing dispersion interactions and subtle conformational energy differences [40]. However, it can overestimate dispersion with certain basis sets, as observed in early hexane conformer studies where MP2/6-311G incorrectly stabilized the ggg conformer over gtg—an error corrected by higher-level CCSD calculations [69].
Density Functional Theory (DFT), specifically the hybrid functional B3LYP, provides a good compromise. Its higher accuracy over HF is attributed to the inclusion of electron correlation via the exchange-correlation functional [70] [71]. Nonetheless, DFT's performance is not uniform and is highly dependent on the chosen functional. DFT errors can be decomposed into functional-driven errors (inherent to the approximate functional) and density-driven errors (arising from an inaccurate electron density) [72]. For systems where self-consistent DFT densities are poor, using the Hartree-Fock density in a non-self-consistent procedure (HF-DFT) can significantly reduce errors, a technique known as density-corrected DFT (DC-DFT) [72].
The Hartree-Fock (HF) method, with its neglect of electron correlation, shows the largest errors among the QM methods (MAE: 0.81-1.00 kcal mol⁻¹). This limitation is particularly pronounced for conformations where electron correlation contributions, such as van der Waals interactions, are non-negligible [40] [71].
Molecular Mechanics Force Fields
Force field performance is directly linked to the quality and breadth of their parameterization.
- Advanced Force Fields (MMFF94, MM3): Their superior performance is a result of being meticulously parameterized against extensive experimental data and high-level QM calculations for organic molecules [40]. For example, the MMFF94 force field was trained on a wide variety of molecular structures, including conformational energies, to achieve chemical accuracy for drug-like compounds [40].
- Generic Force Fields (UFF, DREIDING): These force fields are designed for maximum elemental coverage across the periodic table, often using general rules rather than specific molecule-based parameterization. This generality comes at the cost of accuracy for specific organic systems, leading to larger mean errors [40].
The field is evolving with data-driven approaches like the ByteFF force field, which uses graph neural networks trained on millions of QM-derived molecular fragment geometries and torsion profiles to predict parameters across a expansive chemical space, showing state-of-the-art performance in predicting geometries and conformational energies [73].
The Scientist's Toolkit
For researchers embarking on conformational energy studies, particularly in drug discovery, the following tools and resources are essential.
Table 2: Essential Research Reagents and Computational Tools
| Tool Name | Type | Primary Function in Conformational Analysis |
|---|---|---|
| DLPNO-CCSD(T) | Quantum Chemistry Method | Provides gold-standard reference energies for benchmarking and validating more efficient methods [40]. |
| B3LYP/def2-SVP | Density Functional Theory | Offers a robust and efficient level of theory for geometry optimization and single-point energy calculations [40]. |
| MMFF94 & MM3 | Advanced Force Fields | Enable high-throughput conformational sampling and scoring of drug-like molecules with reasonable accuracy [40]. |
| Graph Neural Networks (GNN) | Machine Learning Architecture | Used in modern force field development (e.g., ByteFF) to predict accurate molecular mechanics parameters across wide chemical spaces [73]. |
| QM/MM | Hybrid Method | Allows accurate modeling of chemical reactions or ligand binding in a biomolecular environment by combining QM and MM regions [70] [71]. |
This quantitative review unequivocally demonstrates a hierarchy of accuracy for predicting conformational energies, with post-HF methods leading in accuracy, followed closely by DFT, and with advanced, well-parameterized force fields providing a highly efficient alternative for high-throughput applications. The choice of method must be guided by the specific requirements of the study, balancing the need for chemical accuracy against available computational resources. The ongoing integration of machine learning with traditional computational chemistry holds the promise of developing next-generation force fields that can approach QM-level accuracy for vastly larger molecular systems, thereby accelerating computational drug discovery and materials design.
Choosing the right computational method is crucial for predicting molecular properties in drug discovery and materials science. This guide provides an objective performance comparison of various quantum mechanical (QM) and molecular mechanics (MM) methods for calculating conformational energies, a key property in computer-aided drug design.
Quantitative Performance at a Glance
The following tables summarize the performance of different methods in reproducing accurate conformational energies, based on a benchmark study of 145 reference organic molecules using DLPNO-CCSD(T) theory as the reference [74].
Quantum Mechanical Methods Performance
| Method | Mean Error vs. Reference (kcal mol⁻¹) | Key Characteristics |
|---|---|---|
| MP2 | 0.35 | Highest accuracy QM method tested; good for electron correlation [74] |
| DFT (B3LYP) | 0.69 | Best balance of accuracy and computational cost [74] |
| Hartree-Fock (HF) | 0.81 - 1.0 | Lacks electron correlation; tends to overestimate bond lengths [74] [59] |
Molecular Mechanics (Force Fields) Performance
| Force Field | Mean Error vs. Reference (kcal mol⁻¹) | Key Characteristics |
|---|---|---|
| MMFF94 | 1.30 | Robust, reliable; widely used in drug design [74] [75] |
| MM3-00 | 1.28 | Top-performing Allinger force field for conformational energies [74] |
| MM4 | 2.05 | Later version but showed higher error in this benchmark [74] |
| MMX / MM+ | 1.77 - 2.01 | MM2-91 clones with moderate performance [74] |
| GAFF/GAFF2 | Varies | Slightly better for QM energies; comparable accuracy for geometries [76] [77] |
| OpenFF (Parsley) | Varies | Modern, approaching state-of-the-art accuracy (e.g., OPLS3e) [76] |
| UFF / DREIDING | ~3.70 | Lowest performance; general-purpose but less accurate for organics [74] |
Detailed Methodologies and Experimental Protocols
The quantitative data presented above stems from a specific, reproducible benchmarking protocol. Understanding this methodology is key to interpreting the results.
Benchmarking Workflow for Conformational Energies
The diagram below outlines the general workflow used to generate the comparative data [74] [76].
Key Experimental Details
- Reference Molecules: The benchmark set included 145 organic molecules, featuring fragments common in drug design, such as hydrocarbons, haloalkanes, and oxygen-, nitrogen-, phosphorus-, and sulphur-containing compounds [74].
- Reference Method: The highly accurate DLPNO-CCSD(T) theory was used to generate the reference conformational energies and barriers. This method accounts for electron correlation with high precision and is considered a reliable source of "true" values when experimental data is unavailable [74].
- Comparison Metric: The primary metric for comparison was the mean absolute error (MAE) in kcal mol⁻¹ between the conformational energy calculated by a method and the reference DLPNO-CCSD(T) value. A lower MAE indicates better performance [74].
The Scientist's Toolkit: Research Reagent Solutions
This table details the key computational "reagents" — the methods and force fields — discussed in this guide, along with their primary functions and considerations for use.
| Tool Name | Type | Primary Function | Key Considerations |
|---|---|---|---|
| MP2 | Quantum Mechanical | Accurate single-point energy calculations, accounting for electron correlation. | High computational cost; can be sensitive to basis set size; recommended for final energy evaluations on DFT-optimized structures [74] [78]. |
| DFT (B3LYP) | Quantum Mechanical | Balanced structure optimization and energy calculation. | Good "default" choice for many applications; hybrid functional that mixes HF and DFT exchange [74] [59]. |
| Hartree-Fock (HF) | Quantum Mechanical | Base-level wavefunction calculation; starting point for post-HF methods. | No electron correlation; low accuracy for conformational energies; fast but largely superseded for property prediction [74]. |
| MMFF94(s) | Molecular Mechanics (Force Field) | High-speed conformational search and scoring in drug discovery. | Fast and robust; parameterized for a wide range of organic molecules; performance is systematically improvable [75] [76]. |
| GAFF/GAFF2 | Molecular Mechanics (Force Field) | Molecular dynamics simulations of small organic molecules. | Slightly better at reproducing QM energies; commonly used in simulation packages like AMBER [76] [77]. |
| OpenFF (Parsley) | Molecular Mechanics (Force Field) | Modern, highly parameterized force field for biomolecular simulation. | SMIRKS-based parameter assignment; shows rapid improvement and is approaching the accuracy of top force fields like OPLS3e [76]. |
Key Insights and Practical Recommendations
- For Highest Accuracy: MP2 is the most accurate method for conformational energies, but its computational expense makes it impractical for large systems or high-throughput virtual screening [74].
- For Best Value: DFT (B3LYP) offers the best compromise, providing good accuracy at a substantially lower computational cost than MP2, making it suitable for geometry optimizations and energy calculations on medium-sized systems [74].
- For High-Throughput Screening: Force Fields are indispensable. MMFF94(s) and modern successors like OPLS3e and OpenFF Parsley provide sufficient accuracy for conformational analysis and docking of thousands of molecules, at a fraction of the QM cost [74] [76] [75].
- Know the Limitations: Hartree-Fock is not recommended for accurate conformational energy predictions due to its lack of electron correlation. Simple general-purpose force fields like UFF and DREIDING should be used with caution for organic molecules, as they show significantly higher errors [74].
In the rigorous fields of computational chemistry and artificial intelligence, the evaluation of model performance often centers on two competing ideals: peak accuracy and robustness. Peak accuracy represents a model's optimal performance under ideal, often narrowly defined, conditions. Robustness, conversely, measures its ability to maintain stable performance when faced with variations, perturbations, or distribution shifts in input data. For researchers and professionals in drug development, this trade-off is particularly acute when selecting computational methods for predicting conformational energies, where the choice between highly specialized and broadly stable models can significantly impact the reliability of virtual screening and property prediction.
This guide objectively compares these trade-offs across both AI and computational chemistry domains, demonstrating that the pursuit of state-of-the-art (SOTA) benchmark performance often comes at the cost of decreased stability. Through structured experimental data and methodological analysis, we provide a framework for interpreting benchmarks that aligns with the practical demands of drug discovery applications, where models must perform reliably across diverse molecular scaffolds and under non-ideal computational conditions.
Defining the Metrics: From AI to Computational Chemistry
Quantifying Robustness in Benchmarking
The concept of robustness extends beyond simple error metrics to encompass a model's sensitivity to input variations. In comprehensive robustness evaluations like the Res-Bench framework for Multimodal Large Language Models (MLLMs), researchers employ multiple specialized metrics to capture different aspects of stability [79]:
- Spearman’s Correlation: Measures the monotonic relationship between input quality (e.g., image resolution) and performance, where strong positive correlation indicates sensitivity to degradation.
- Absolute Continuous Error (ACE) and Relative Continuous Error (RCE): Quantify performance volatility across different input resolution levels, with lower values indicating greater stability.
- Stability Metrics for Molecular Methods: In computational chemistry, robustness is often measured as the variance in error across diverse molecular classes or as mean absolute error (MAE) consistency across different conformational energy calculations.
The Peak Accuracy Illusion in Leaderboards
Benchmark leaderboards often prioritize peak accuracy metrics that can be misleading for real-world applications. Studies indicate that leaderboard rankings can shift up to eight positions through minor changes to evaluation formats, and identical models submitted under different names have received discrepancies of up to 17 points on standardized evaluations [80]. This volatility underscores how peak accuracy metrics can create a false sense of performance superiority that may not translate to applied settings.
Experimental Evidence of the Trade-Off
Domain-Specific Trade-offs: A Comparative Analysis
Multimodal AI Performance Patterns
In MLLM evaluations, a clear architectural trade-off emerges between models designed for peak performance versus robustness. When tested across 12 resolution levels and 6 capability dimensions comprising 14,400 samples, researchers found that native dynamic processing methods tend to achieve higher peak performance but are less robust to resolution changes, while patch-based methods demonstrate better robustness but at the cost of lower overall performance [79]. This pattern held across both proprietary models (GPT-4o, Gemini-1.5 Pro) and open-source models (LLaVA-OneVision, Qwen2.5-VL), suggesting a fundamental architectural constraint.
Table 1: Robustness-Accuracy Trade-off in MLLM Architectures
| Architecture Type | Peak Accuracy | Robustness (ACE) | Resolution Stability | Best Use Case |
|---|---|---|---|---|
| Native Dynamic Processing | High (~89%) | Low (High ACE) | Sensitive to degradation | Controlled environments with quality inputs |
| Patch-Based Methods | Moderate (~82%) | High (Low ACE) | Maintains performance across resolutions | Real-world applications with variable input quality |
Computational Chemistry Method Comparisons
In conformational energy calculations for drug-like molecules, similar trade-offs appear between computational methods. When benchmarking common efficient computational methods against coupled cluster theory (DLPNO-CCSD(T)) using 158 conformational energies of 145 reference organic molecules, researchers observed distinct accuracy-robustness patterns [40]:
Table 2: Performance-Robustness Trade-offs in Conformational Energy Calculations
| Computational Method | Mean Error vs DLPNO-CCSD(T) (kcal mol⁻¹) | Computational Cost | Robustness Across Molecular Classes | Typical Application Context |
|---|---|---|---|---|
| MP2 | 0.35 | Very High | High | Gold-standard reference calculations |
| B3LYP | 0.69 | High | Moderate-High | Research-grade screening |
| HF/6-31G* | 0.81-1.0 | Moderate | Moderate | Initial conformational sampling |
| MMFF94 | 1.30 | Low | Moderate | High-throughput virtual screening |
| MM3-00 | 1.28 | Low | High | Ligand conformation analysis |
| UFF | 3.77 | Very Low | Low | Initial structure optimization |
The data reveals that higher accuracy methods (MP2, B3LYP) achieve superior peak performance but require substantial computational resources, limiting their application in large-scale virtual screening. Conversely, molecular mechanics methods (MMFF94, MM3-00) offer greater practical robustness and efficiency with moderate accuracy compromises, making them suitable for high-throughput applications [40].
Real-World Performance vs. Benchmark Performance
Controlled studies demonstrate how benchmark performance often fails to predict real-world utility. In a randomized controlled trial with experienced open-source developers, researchers found that AI tools that performed impressively on coding benchmarks actually slowed developers down by 19% on realistic software development tasks [81]. This performance gap was attributed to benchmark tasks being "well-scoped, algorithmically scorable tasks" that don't capture the nuanced requirements, implicit knowledge, and quality standards of real-world applications.
Similarly, in drug discovery applications, methods that excel on standardized benchmark datasets may underperform when applied to novel molecular scaffolds or under different simulation conditions, highlighting the importance of robustness across chemical space.
Methodological Framework for Balanced Evaluation
Experimental Protocols for Comprehensive Assessment
To properly evaluate both peak accuracy and robustness in computational methods, researchers should implement a stratified assessment protocol:
Multi-Fidelity Validation Protocol
- Primary Accuracy Assessment: Measure performance against high-fidelity reference data (e.g., DLPNO-CCSD(T) for conformational energies) on standardized benchmark molecules.
- Perturbation Testing: Introduce controlled variations in input quality, molecular structure, or computational parameters to assess stability.
- Cross-Domain Validation: Test methods on diverse molecular classes beyond the training or optimization dataset.
- Resource Efficiency Tracking: Monitor computational requirements relative to accuracy gains to determine practical utility.
Workflow for Balanced Benchmark Design
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Reagents for Robustness-Accuracy Evaluation
| Research Reagent | Function | Application Context |
|---|---|---|
| DLPNO-CCSD(T)/cc-pVTZ Reference Data | Provides benchmark-quality conformational energies for 145+ organic molecules | Gold-standard validation for method development [40] |
| QMugs Dataset | Quantum mechanical properties for ~665k biologically relevant molecules | Large-scale validation of computational methods [40] |
| Res-Bench Framework | Comprehensive robustness metrics (ACE, RCE, Spearman correlation) | Quantifying model stability across input variations [79] |
| Multi-Fidelity Validation Set | Curated molecules with experimental and high-level computational data | Cross-validation of methods across accuracy domains |
| MMFF94/MM3 Force Fields | Efficient conformational sampling with validated parameters | Baseline comparison for new method development [40] |
Implications for Method Selection in Drug Discovery
The trade-off between robustness and peak accuracy necessitates context-dependent method selection in drug development pipelines:
Targeted High-Accuracy Applications For final-stage lead optimization or precise conformational energy ranking, higher-cost methods like MP2 or DLPNO-CCSD(T) provide the necessary accuracy despite computational intensity [40]. These methods are essential for establishing reliable reference data and validating more efficient approaches.
High-Throughput Screening Applications In early-stage virtual screening or conformational sampling across large compound libraries, robust molecular mechanics methods (MMFF94, MM3-00) offer the best balance of reasonable accuracy and computational efficiency [40]. Their stability across diverse molecular structures makes them suitable for exploring unknown chemical space.
Hybrid Approaches Multi-level strategies that combine efficient filtering using robust methods with refined analysis using high-accuracy methods provide an optimal balance. For example, using MMFF94 for initial conformational sampling followed by DFT refinement on promising candidates leverages the strengths of both approaches.
The pursuit of state-of-the-art performance on standardized benchmarks must be tempered with rigorous evaluation of robustness across diverse conditions. As evidenced by studies in both AI and computational chemistry, methods optimized for peak accuracy frequently exhibit sensitivity to input variations and computational parameters that limit their real-world utility.
For researchers and professionals in drug development, this necessitates a shift in evaluation criteria—from singular focus on leaderboard rankings to comprehensive assessment of performance stability across chemically diverse space. By implementing the multi-fidelity validation protocols and balanced metrics outlined in this guide, the field can develop computational methods that excel not only in controlled benchmarks but also in the demanding, variable environments of practical drug discovery.
For decades, density functional theory (DFT) has been a cornerstone of computational chemistry, providing insights into electronic structures for drug discovery and materials science. However, the accuracy of traditional DFT is limited by approximations in the exchange-correlation functional, often resulting in errors of 2-3 kcal·mol⁻¹ for many molecules, which is insufficient for reliable predictive modeling in drug development [82]. The gold-standard coupled-cluster theory, such as CCSD(T), provides the required quantum chemical accuracy but at a computational cost that prohibits its application to large systems or long molecular dynamics simulations [82].
Machine learning (ML) is now revolutionizing this field by enabling the development of novel density functionals that bridge this accuracy-speed gap. These ML-driven approaches learn from high-quality reference data to predict electronic properties, energies, and forces with unprecedented accuracy while maintaining computational feasibility. This guide explores and compares these emerging validated alternatives, focusing on their performance for calculating conformational energies—a critical property in computer-aided drug design where precise energy rankings determine molecular stability and activity.
Core Approaches and Theoretical Foundations
2.1.1 Direct DFT Emulation and Learning from the Random-Phase Approximation Several innovative frameworks have been developed to emulate traditional DFT at a fraction of the computational cost. One prominent approach uses deep learning to map atomic structures directly to electronic charge densities and other properties in an end-to-end manner, successfully bypassing the explicit solution of the Kohn-Sham equation with orders of magnitude speedup while maintaining chemical accuracy [83]. This method employs atomic fingerprints to represent structural and chemical environments, then uses deep neural networks to predict electronic charge density descriptors, which are subsequently used to compute other properties like total energy, atomic forces, and stress tensors [83].
Another significant approach involves mapping the random-phase approximation (RPA)—a high-level functional from the top of Jacob's ladder—to a pure Kohn-Sham density functional using machine learning [84]. The resulting ML-RPA model acts as a nonlocal extension of the standard generalized gradient approximation (GGA), using density descriptors that are nonlocal counterparts of the local density and its gradient [84]. This approach incorporates derivative information through RPA optimized effective potentials during training, similar to how atomic forces improve machine-learned force fields [84].
2.1.2 Δ-Learning and Hybrid Strategies A particularly efficient strategy known as Δ-DFT involves learning only the correction to a standard DFT calculation rather than the total energy itself [82]. This approach recognizes that the error in DFT is more amenable to learning than the total energy itself, significantly reducing the amount of training data required to achieve quantum chemical accuracy [82]. By exploiting molecular point group symmetries, this method can drastically reduce the required training data while enabling the extraction of CCSD(T) energies from standard DFT calculations with essentially no additional cost beyond the initial generation of training data [82].
Key Architectural Components
Descriptors and Representations: Machine-learning density functionals rely heavily on sophisticated descriptors of atomic and electronic environments. The atom-centered AGNI fingerprints represent structural and chemical environments in a translation, permutation, and rotation invariant manner [83]. For representing electronic densities, approaches include grid-based schemes, atom-based representations using Gaussian-type orbitals (GTOs), and power spectrum representations of atomic environments adapted for electronic densities [83] [84].
Learning Algorithms: Kernel ridge regression (KRR) with Gaussian kernels has been successfully employed for learning exchange-correlation functionals [84] [82]. Deep neural networks (DNNs) form the backbone of more complex frameworks such as DeePHF and DeePKS, which are specifically tailored for drug-like molecules [50] [83].
Performance Comparison and Experimental Validation
Accuracy Assessment on Conformational Energies
The critical test for any electronic structure method in drug development is its performance on conformational energies, where small energy differences determine molecular stability and biological activity. The table below summarizes the performance of various computational methods against the DLPNO-CCSD(T) benchmark for 158 conformational energies of organic molecules representative of drug fragments [40].
Table 1: Performance comparison of computational methods for conformational energies
| Method Category | Specific Method | Mean Error vs DLPNO-CCSD(T) (kcal·mol⁻¹) | Computational Cost |
|---|---|---|---|
| Post-HF Methods | DLPNO-CCSD(T) (reference) | 0.00 (by definition) | Very High |
| MP2 | 0.35 | High | |
| Traditional DFT | B3LYP | 0.69 | Medium |
| Wavefunction Methods | HF (with different basis sets) | 0.81-1.00 | Medium-High |
| Machine Learning DFT | DeePHF/DeePKS | ~Chemical accuracy (exact value not specified) | Low (after training) |
| ML-DFT (general) | Chemical accuracy achieved [83] | Low (after training) | |
| Δ-DFT | <1.0 (quantum chemical accuracy) [82] | Low (after training) | |
| Force Fields | MM3-00 | 1.28 | Very Low |
| MMFF94 | 1.30 | Very Low | |
| MM3-96 | 1.40 | Very Low | |
| MMX | 1.77 | Very Low | |
| MM+ | 2.01 | Very Low | |
| MM4 | 2.05 | Very Low | |
| DREIDING | 3.63 | Very Low | |
| UFF | 3.77 | Very Low |
Machine learning density functionals demonstrate exceptional performance, achieving chemical accuracy (typically defined as errors below 1 kcal·mol⁻¹) that rivals or surpasses traditional quantum chemistry methods while operating at computational costs closer to force fields after the initial training phase [83] [82]. Specific ML-DFT approaches like DeePHF and DeePKS, trained on datasets labeled at the CCSD(T)/def2-TZVP level, have achieved chemical accuracy for molecular energies with excellent transferability [50].
Specialized Benchmarks for Drug-like Molecules
For drug-like molecules, the Δ-DFT method has shown remarkable robustness, correctly capturing conformational changes and energy barriers where standard DFT fails [82]. This capability was highlighted in molecular dynamics simulations of resorcinol (C6H4(OH)₂), where Δ-DFT successfully corrected DFT-based trajectories to obtain coupled-cluster accuracy, even for strained geometries and conformer changes [82]. This demonstrates the potential of ML density functionals to enable reliable gas-phase MD simulations with quantum chemical accuracy, which is essential for predicting drug behavior in physiological environments.
Experimental Protocols and Methodologies
Reference Data Generation and Training
4.1.1 High-Quality Reference Datasets The development of accurate ML density functionals relies on high-quality training data derived from advanced quantum chemical methods:
- CCSD(T) Calculations: Considered the gold standard, provided reference energies for organic molecules and drug fragments in multiple studies [82] [40]. The DLPNO-CCSD(T) approximation enables calculations on larger systems while maintaining high accuracy [40].
- RPA Reference Data: Used for training ML-RPA functionals, incorporating both RPA exchange-correlation energies and derivative information in the form of RPA optimized effective potentials [84].
Large-scale datasets have been crucial for training and validation. The PubChemQCR dataset, for instance, contains approximately 3.5 million relaxation trajectories and over 300 million molecular conformations computed at various levels of theory, with each conformation labeled with total energy and atomic forces [85]. Similarly, the QMugs dataset comprises quantum mechanical properties for over 665,000 biologically relevant molecules extracted from the ChEMBL database [40].
4.1.2 Training Procedures and Protocols
- DeePHF/DeePKS Training: These models were trained with limited datasets labeled at the CCSD(T)/def2-TZVP level, demonstrating the ability to achieve chemical accuracy with relatively small but high-quality training data [50].
- ML-DFT Framework Training: This approach used over 118,000 structures of organic molecules, polymer chains, and polymer crystals containing C, H, N, and O atoms, divided into training, validation, and test sets with a 90:10 split, and the training set further split 80:20 between training and validation [83].
- Δ-DFT Training: This method learns the difference between DFT and CCSD(T) energies as a functional of input DFT densities, requiring significantly less training data than learning either DFT or CCSD(T) energies separately [82].
Validation Methodologies
4.2.1 Conformational Energy Validation Comprehensive validation against established benchmarks is crucial. The performance of ML density functionals has been assessed against:
- 158 Conformational Energies and Barriers: A curated set of reference organic molecules that include model fragments used in computer-aided drug design [40].
- Specialized Test Sets: Including the 37conf8 database of 37 organic molecules representing pharmaceuticals, drugs, catalysts, and industry-related chemicals [40].
- Molecular Dynamics Simulations: Validating the ability to produce correct dynamics and energy barriers during conformational changes [82].
4.2.2 Transferability Assessment A critical test for ML density functionals is their performance on systems not included in the training data:
- Size Transferability: Evaluating performance on larger systems than those in the training set [83].
- Chemical Space Transferability: Assessing accuracy across diverse molecular structures and elements [50] [83].
- Surface and Interface Applications: Testing performance for challenging cases like diamond surfaces, where ML-RPA reached the accuracy of state-of-the-art van der Waals functionals [84].
Workflow and Signaling Pathways
The application of machine learning density functionals follows a systematic workflow from data preparation to validation. The diagram below illustrates this process, highlighting the key stages and decision points in developing and applying these methods.
Table 2: Essential research reagents and resources for ML-DFT development
| Resource Category | Specific Resource | Function and Application | Key Features |
|---|---|---|---|
| Reference Datasets | PubChemQCR | Provides relaxation trajectories for training and validation | ~3.5M trajectories, 300M+ conformations with energy/force labels [85] |
| QMugs | Quantum properties of drug-like molecules | 665k+ molecules from ChEMBL, DFT densities [40] | |
| QM7-X | Comprehensive dataset of molecular structures | ~4.2M structures, includes force labels [85] | |
| Software Packages | DeePHF/DeePKS | Specialized ML-DFT for drug-like molecules | Achieves chemical accuracy for molecular energies [50] |
| ML-DFT Framework | End-to-end DFT emulation | Bypasses Kohn-Sham equations with orders of magnitude speedup [83] | |
| VASP | First-principles calculations | Generates reference data for ML model training [83] | |
| Reference Methods | DLPNO-CCSD(T) | Gold-standard reference method | Provides accurate conformational energies for benchmarking [40] |
| RPA with OEP | High-level functional for training | Provides energies and potentials for ML-RPA training [84] | |
| Descriptors | AGNI Atomic Fingerprints | Represents atomic environments | Translation, permutation, and rotation invariant structural descriptors [83] |
| Power Spectrum Descriptors | Represents electronic densities | Nonlocal extension of GGA ingredients for ML-DFT [84] |
Machine-learning density functionals represent a paradigm shift in computational chemistry, offering quantum chemical accuracy at computational costs that enable practical applications in drug discovery and development. These emerging validated alternatives have demonstrated exceptional performance in calculating conformational energies—a critical property for predicting molecular behavior in drug design. While challenges remain in ensuring broad transferability across diverse chemical spaces, the rapid advancements in this field suggest that ML-driven quantum chemistry methods will soon become indispensable tools for pharmaceutical research. As dataset size and diversity continue to grow, and as model architectures become more sophisticated, these approaches promise to unlock new possibilities for predictive molecular modeling with unprecedented accuracy and efficiency.
Conclusion
The reliable prediction of conformational energies is non-negotiable for confident decision-making in computational drug discovery. This guide synthesizes key takeaways, affirming that while DLPNO-CCSD(T) provides the most accurate reference, method selection must be guided by the specific task. MP2 emerges as a top-performing post-HF method for accuracy, with modern, dispersion-corrected DFT functionals like r2SCAN-3c offering a robust and efficient balance for day-to-day applications. Force fields, while fast, show significantly higher errors and require careful validation for drug-like molecules. Looking forward, the integration of machine learning potentials trained on high-quality QM data sets promises to revolutionize the field, offering near-quantum accuracy at dramatically reduced computational cost. By adopting the validated protocols and comparative frameworks outlined here, researchers can significantly enhance the predictive power of their molecular modeling, ultimately accelerating the discovery of new therapeutics.
References
- 1. Searching for bioactive conformations of drug-like ligands with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5432473/]
- 2. Assessing the performance of OMEGA with respect to ... [https://www.sciencedirect.com/science/article/abs/pii/S1093326302002048]
- 3. GEOM, energy-annotated molecular conformations for ... [https://www.nature.com/articles/s41597-022-01288-4]
- 4. dimers of polycyclic (hetero-)aromatics [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp01010a]
- 5. The Role of Exchange Energy in Modeling Core-Electron ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12251301/]
- 6. A diverse and chemically relevant solvation model benchmark ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d5sc06406f]
- 7. A comparison of DLPNO-CCSD(T) and CCSD(T) method ... [https://www.sciencedirect.com/science/article/abs/pii/S2210271X20302346]
- 8. Machine-learning interatomic potentials achieving CCSD(T ... [https://arxiv.org/html/2508.14306v1]
- 9. Conformational energies of reference organic molecules [https://pmc.ncbi.nlm.nih.gov/articles/PMC10618395/]
- 10. Assessment of the DLPNO Binding Energies of Strongly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7144154/]
- 11. Reliable Structures and Electronic Energies of Small Water ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12516732/]
- 12. Revisiting Boron Cluster Dihydrogen Bonding: Improved ... [https://chemrxiv.org/engage/chemrxiv/article-details/68a8628523be8e43d6833304]
- 13. Fragment‐based drug discovery—the importance of high ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9627785/]
- 14. Fragment-based drug discovery: A graphical review [https://www.sciencedirect.com/science/article/pii/S2590257125000215]
- 15. The QDπ dataset, training data for drug-like molecules and ... [https://www.nature.com/articles/s41597-025-04972-3]
- 16. Best‐Practice DFT Protocols for Basic Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9826355/]
- 17. Fragment-based drug discovery: opportunities for organic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8130625/]
- 18. Fragment Based Drug Discovery [https://www.drugdiscoverychemistry.com/Fragment-Based-Drug-Discovery/16]
- 19. Evaluation of quantum chemistry calculation methods for ... [https://pubs.rsc.org/en/content/articlehtml/2023/ra/d3ra06783a]
- 20. “DompeKeys”: a set of novel substructure-based descriptors ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00813-4]
- 21. Comparing the performance of density functionals in ... [https://www.sciencedirect.com/science/article/abs/pii/S0039602820303125]
- 22. DFT based structural modeling of chemotherapy drugs via ... [https://www.nature.com/articles/s41598-025-97982-5]
- 23. Comparison of the Performance of Density Functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10146789/]
- 24. Application of Density Functional Theory to Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11989887/]
- 25. Predicting the Post-Hartree-Fock Electron Correlation ... [https://www.mdpi.com/1420-3049/30/17/3500]
- 26. Performance of DFT functionals for properties of small ... [https://www.sciencedirect.com/science/article/pii/S2352179120300077]
- 27. a validation study of catalysts for olefin metathesis and ... [https://pubs.rsc.org/en/content/articlelanding/2012/dt/c2dt12232d]
- 28. A Robust Crystal Structure Prediction Method to Support ... [https://chemrxiv.org/engage/chemrxiv/article-details/676a0ce6fa469535b999990a]
- 29. Energy landscapes of functional proteins are inherently risky [https://pmc.ncbi.nlm.nih.gov/articles/PMC4416114/]
- 30. Energy landscape of conformational changes for a single ... [https://www.nature.com/articles/s44328-024-00014-x]
- 31. Composite Methods in Quantum Chemistry - Corin Wagen [https://corinwagen.github.io/public/blog/20230922_composite.html]
- 32. Basis set — BAND 2025.1 documentation - SCM [https://www.scm.com/doc/BAND/Accuracy_and_Efficiency/Basis_Set.html]
- 33. Quantum chemistry composite methods [https://en.wikipedia.org/wiki/Quantum_chemistry_composite_methods]
- 34. A Comparison of the Behavior of Functional/Basis Set ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3073166/]
- 35. Methods and tools for assessing the accuracy of composite ... [https://www.sciencedirect.com/science/article/abs/pii/S2352492823026478]
- 36. Reliability of ab initio (HF, post HF and DFT) methods and ... [https://www.sciencedirect.com/science/article/abs/pii/S0166128002000386]
- 37. opportunities for innovation in drug discovery - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12641420/]
- 38. Computational approaches streamlining drug discovery [https://www.nature.com/articles/s41586-023-05905-z]
- 39. Single point energies - ORCA 6.0 TUTORIALS [https://www.faccts.de/docs/orca/6.0/tutorials/prop/single_point.html]
- 40. Conformational energies of reference organic molecules [https://link.springer.com/article/10.1007/s10822-023-00513-5]
- 41. 4.2 Hartree–Fock Calculations [https://manual.q-chem.com/4.3/sect0037.html]
- 42. XTBDFT: Automated workflow for conformer searching of ... [https://www.sciencedirect.com/science/article/pii/S2352711022001601]
- 43. PyConSolv: A Python Package for Conformer Generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10498442/]
- 44. An efficient workflow for generation of conformational ensembles of... [https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp01010a]
- 45. An Efficient Workflow for Generation of Conformational ... [https://chemrxiv.org/engage/chemrxiv/article-details/67c39135fa469535b985ff55]
- 46. analysis of GFN Comparative in geometry optimization of... methods [https://arxiv.org/html/2505.09606v1]
- 47. Dispersion-correction density functional theory (DFT+D) ... [https://www.sciencedirect.com/science/article/abs/pii/S2352214322001265]
- 48. Optimizing the basis set extrapolation parameter for weak ... [https://www.sciencedirect.com/science/article/abs/pii/S2210271X25004311]
- 49. Assessing Density Functionals and Dispersion Corrections for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12511928/]
- 50. A machine learning-based high-precision density ... [https://www.sciencedirect.com/science/article/pii/S2949747723000374]
- 51. Benchmarking simulacra AI's Quantum Accurate Synthetic ... [https://arxiv.org/html/2511.07433v1]
- 52. Digitized material design and performance prediction ... [https://www.frontiersin.org/journals/materials/articles/10.3389/fmats.2025.1599439/full]
- 53. Balancing accuracy and efficiency: co-design of hybrid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570339/]
- 54. Essential guidelines for computational method benchmarking [https://pmc.ncbi.nlm.nih.gov/articles/PMC6584985/]
- 55. Statistical analysis in method comparison studies part one [https://acutecaretesting.org/en/articles/statistical-analysis-in-method-comparison-studies-part-one]
- 56. Concise Experimental and DFT Study of Newly ... [https://www.sciencedirect.com/science/article/pii/S2667022425001513]
- 57. Best Statistical Tools for Data Analysis and Visualization [https://www.6sigma.us/six-sigma-in-focus/statistical-tools/]
- 58. Density functional benchmark for quadruple hydrogen bonds [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d5cp00836k]
- 59. The "Charlotte's Web" of Density-Functional Theory (DFT) [https://rowansci.com/publications/the-charlottes-web-of-dft]
- 60. Comparisons of different force fields in conformational ... [https://www.sciencedirect.com/science/article/abs/pii/S0040402020311236]
- 61. A Benchmark for Quantum Chemistry Relaxations via ... [https://arxiv.org/html/2506.23008v2]
- 62. Density Functionals (XC) — ADF 2025.1 documentation - SCM [https://www.scm.com/doc/ADF/Input/Density_Functional.html]
- 63. Performance of quantum chemistry methods for a ... [https://www.sciencedirect.com/org/science/article/pii/S2041652024018029]
- 64. Theoretical study on conformational of transition metal... energies [https://pubs.rsc.org/en/content/articlehtml/2021/cp/d0cp04696e]
- 65. Comparing ANI-2x, ANI-1ccx neural networks, force field, ... [https://www.nature.com/articles/s41598-024-62242-5]
- 66. Customized force parameters using a hybrid field //GFN2-xTB... DFT [https://cresset-group.com/about/news/customized-force-field-parameters-dftgfn2-xtb/]
- 67. Multi-level QTAIM-enriched graph neural networks for ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00220f]
- 68. 4.3 Density Functional Theory [https://manual.q-chem.com/4.3/sect-DFT.html]
- 69. An ab-initio study of the relative stability of the ggg and ... [https://www.sciencedirect.com/science/article/abs/pii/S0009261499009264]
- 70. Advancing predictive modeling in computational chemistry ... [https://link.springer.com/article/10.1007/s44371-025-00363-0]
- 71. Quantum Mechanics in Drug Discovery - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12249871/]
- 72. Analyzing density-driven errors: Principles and pitfalls [https://chemrxiv.org/engage/chemrxiv/article-details/68d100fa3e708a764999322a]
- 73. Data-driven parametrization of molecular mechanics force ... [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc06640e]
- 74. Conformational energies of reference organic molecules [https://pubmed.ncbi.nlm.nih.gov/37597063/]
- 75. Accuracy evaluation and addition of improved dihedral ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-019-0371-6]
- 76. Benchmark assessment of molecular geometries and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7863993/]
- 77. Benchmark Assessment of Molecular Geometries and ... [https://chemrxiv.org/engage/chemrxiv/article-details/60c74ebfbdbb899d58a39c23]
- 78. Which is better for binding energies: DFT-D3 or RI-MP2? [https://mattermodeling.stackexchange.com/questions/11460/which-is-better-for-binding-energies-dft-d3-or-ri-mp2]
- 79. Res-Bench: Benchmarking the Robustness of Multimodal ... [https://arxiv.org/html/2510.16926v3]
- 80. LLM Benchmarking Decoded: Updates In May 2025 [https://empathyfirstmedia.com/llm-benchmarking-decoded-updates-in-may-2025/]
- 81. Measuring the Impact of Early-2025 AI on Experienced ... [https://metr.org/blog/2025-07-10-early-2025-ai-experienced-os-dev-study/]
- 82. Quantum chemical accuracy from density functional ... [https://www.nature.com/articles/s41467-020-19093-1]
- 83. A deep learning framework to emulate density functional ... [https://www.nature.com/articles/s41524-023-01115-3]
- 84. Machine Learning Density Functionals from the Random ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10601474/]
- 85. A Benchmark for Quantum Chemistry Relaxations via ... [https://arxiv.org/html/2506.23008v1]