Quantum Computing for Molecular Ground States: From Theory to Drug Discovery Applications
This article provides a comprehensive overview of how quantum computing is revolutionizing the search for molecular Hamiltonian ground states, a cornerstone of accurate quantum chemistry.
Quantum Computing for Molecular Ground States: From Theory to Drug Discovery Applications
Abstract
This article provides a comprehensive overview of how quantum computing is revolutionizing the search for molecular Hamiltonian ground states, a cornerstone of accurate quantum chemistry. Aimed at researchers, scientists, and drug development professionals, it explores the foundational principles that give quantum computers a potential edge, details cutting-edge hybrid algorithms already in use on current hardware, and discusses strategies to overcome noise and errors. Furthermore, it examines the emerging evidence of verifiable quantum advantage and compares quantum approaches with classical methods, concluding with an analysis of the transformative future impact on biomedical research and therapeutic discovery.
The Quantum Promise: Why Qubits Can Solve Intractable Chemistry Problems
The determination of the molecular ground state—the lowest energy state of a molecule's electrons—represents one of the most fundamental challenges in quantum chemistry and materials science. This ground state energy and its associated wavefunction govern molecular structure, stability, reactivity, and physical properties, making its accurate computation essential for drug discovery, materials design, and chemical reaction modeling [1]. The molecular Hamiltonian, which encapsulates the total energy of the electrons and nuclei within a molecule, serves as the starting point for this quantum mechanical description [1].
In the context of quantum computing, the molecular Hamiltonian ground state problem has emerged as a potential benchmark application where quantum algorithms may demonstrate significant advantage over classical computational methods. The search for quantum advantage in this domain represents an active frontier of research, bridging quantum chemistry, computational physics, and quantum information science [2]. This technical guide examines the core challenge from both theoretical and computational perspectives, with particular emphasis on emerging quantum approaches.
Theoretical Foundation: The Molecular Hamiltonian
Components of the Coulomb Hamiltonian
The full molecular Hamiltonian, often referred to as the Coulomb Hamiltonian, comprises five distinct physical contributions that collectively describe the energy landscape of the molecular system [1]:
- Nuclear kinetic energy: ( \hat{T}n = -\sumi \frac{\hbar^2}{2Mi}\nabla{\mathbf{R}_i}^2 )
- Electronic kinetic energy: ( \hat{T}e = -\sumi \frac{\hbar^2}{2me}\nabla{\mathbf{r}_i}^2 )
- Electron-nucleus attraction: ( \hat{U}{en} = -\sumi \sumj \frac{Zi e^2}{4\pi \varepsilon0 |\mathbf{R}i - \mathbf{r}_j|} )
- Electron-electron repulsion: ( \hat{U}{ee} = \frac{1}{2}\sumi \sum{j \neq i} \frac{e^2}{4\pi \varepsilon0 |\mathbf{r}i - \mathbf{r}j|} )
- Nuclear-nuclear repulsion: ( \hat{U}{nn} = \frac{1}{2}\sumi \sum{j \neq i} \frac{Zi Zj e^2}{4\pi \varepsilon0 |\mathbf{R}i - \mathbf{R}j|} )
The complexity arises from the coupled nature of these terms, particularly the electron-electron repulsion that creates a many-body problem requiring approximate solutions for all but the smallest molecular systems.
The Born-Oppenheimer Approximation
Practical computation almost universally employs the Born-Oppenheimer approximation, which separates nuclear and electronic motion based on the significant mass disparity between these components [1]. This simplification allows chemists to focus on the electronic Hamiltonian with fixed nuclear positions, generating potential energy surfaces that govern nuclear motion. The electronic Schrödinger equation within this framework becomes:
( \hat{H}{electronic} = \hat{T}e + \hat{U}{en} + \hat{U}{ee} )
where the nuclear-nuclear repulsion term ( \hat{U}_{nn} ) adds a constant energy offset for fixed nuclear configurations.
Classical Computational Approaches
Hierarchy of Methods
Classical computational chemistry employs a spectrum of methods balancing accuracy and computational cost, as summarized in Table 1.
Table 1: Classical Computational Methods for Molecular Ground State Determination
| Method | Key Approach | Scaling Complexity | Key Limitations |
|---|---|---|---|
| Hartree-Fock (HF) | Mean-field approximation | ( \mathcal{O}(N^4) ) | Neglects electron correlation |
| Configuration Interaction (CI) | Linear expansion in Slater determinants | ( \mathcal{O}(N^{6-10}) ) | Size inconsistency; factorial scaling |
| Coupled Cluster (CC) | Exponential ansatz (e.g., CCSD(T)) | ( \mathcal{O}(N^7) ) for CCSD(T) | High polynomial scaling |
| Full CI (FCI) | Exact solution within basis set | Factorial in electrons/orbitals | Limited to very small systems |
| Density Functional Theory (DFT) | Electron density functional | ( \mathcal{O}(N^3) ) | Functional approximation error |
| Quantum Monte Carlo (QMC) | Stochastic sampling | ( \mathcal{O}(N^{3-4}) ) | Fermionic sign problem |
| DMRG/MPS | Tensor network compression | ( \mathcal{O}(N) ) for 1D systems | Accuracy depends on entanglement |
The Full Configuration Interaction Limit
Full Configuration Interaction (FCI) represents the exact numerical solution of the electronic Schrödinger equation within a given one-electron basis set [3]. The FCI wavefunction is expressed as a linear combination of all possible Slater determinants:
( \Psi{\text{FCI}} = c0 |\Psi0\rangle + \sum{i,a} ci^a |\Psii^a\rangle + \sum{i>j,a>b} c{ij}^{ab} |\Psi_{ij}^{ab}\rangle + \cdots )
where ( |\Psi0\rangle ) is the Hartree-Fock determinant, ( |\Psii^a\rangle ) are singly-excited determinants, and higher terms include up to N-tuple excitations for an N-electron system [3]. While FCI provides the benchmark against which all approximate methods are judged, its factorial scaling with system size limits practical application to small molecules with minimal basis sets.
The Challenge of Strong Correlation
Particularly challenging cases for classical methods include systems with strong electron correlation, such as open-shell molecules, transition metal complexes (e.g., iron-sulfur clusters in nitrogenase), and bond dissociation processes [3] [2]. In these scenarios, single-reference methods like Hartree-Fock and standard coupled cluster approximations break down, requiring more sophisticated multi-configurational approaches such as Complete Active Space Self-Consistent Field (CASSCF) methods.
Quantum Computing Approaches
Quantum Algorithm Frameworks
Quantum computing offers potentially transformative approaches to the molecular ground state problem, with several algorithmic frameworks currently under development, as summarized in Table 2.
Table 2: Quantum Algorithms for Molecular Ground State Determination
| Algorithm | Key Principle | Resource Requirements | Current Status |
|---|---|---|---|
| Variational Quantum Eigensolver (VQE) | Hybrid quantum-classical optimization | Shallow circuits; noise-resistant | Demonstrated on small molecules [4] [5] [6] |
| Quantum Phase Estimation (QPE) | Coherent energy measurement | Deep circuits; error correction | Fault-tolerant requirement |
| Quantum Imaginary Time Evolution (QITE) | Non-unitary evolution via parameterization | Moderate circuit depth | Experimental demonstrations [7] [8] |
| Dissipative Lindblad Dynamics | Open quantum systems approach | State preparation via dissipation | Theoretical proposal [9] |
| Quantum Annealing | Adiabatic ground state preparation | Specialized hardware | Hamiltonian reduction applications [3] |
The VQE Framework and Enhancements
The Variational Quantum Eigensolver (VQE) has emerged as a leading algorithm for the Noisy Intermediate-Scale Quantum (NISQ) era due to its relative resilience to noise and minimal quantum resource requirements [4] [6]. VQE employs a hybrid quantum-classical approach where:
- A parameterized quantum circuit (ansatz) prepares trial wavefunctions on a quantum processor
- The expectation value of the molecular Hamiltonian is measured
- A classical optimizer adjusts circuit parameters to minimize the energy
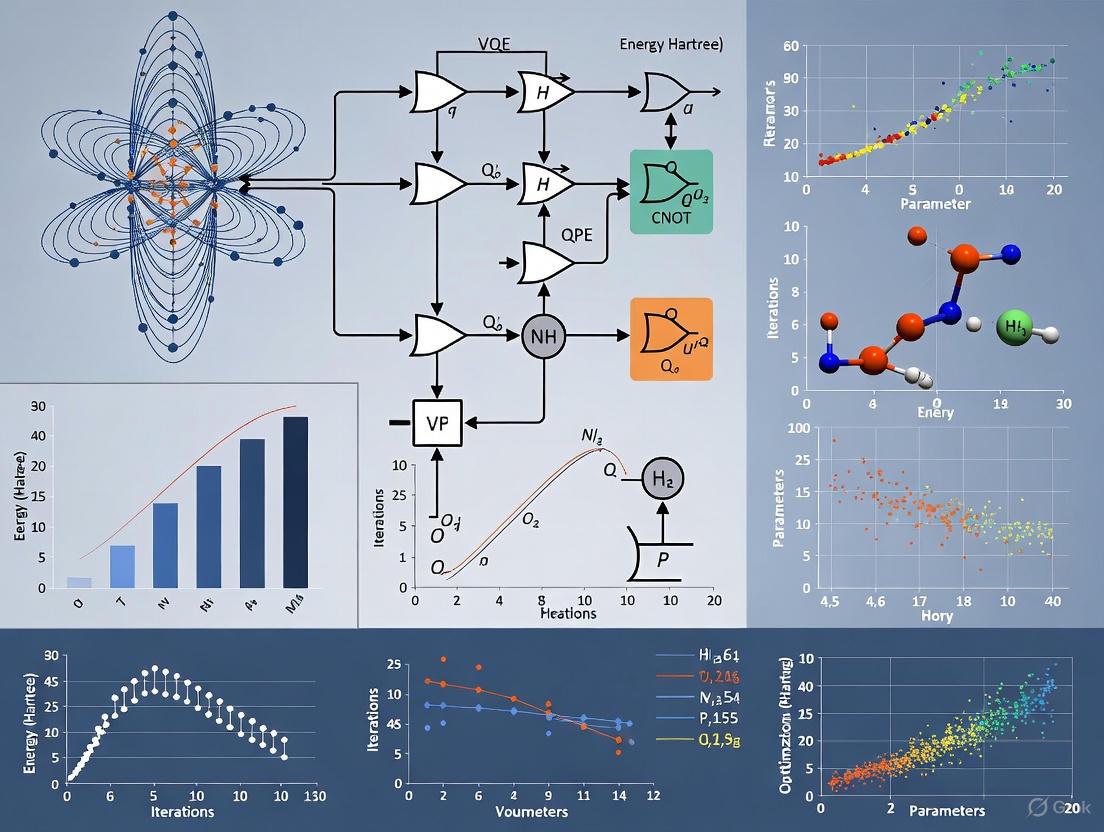
Recent enhancements include adaptive ansatz construction protocols like ADAPT-VQE and K-ADAPT-VQE, which dynamically build problem-specific circuits by selecting operators from a predefined pool based on their gradient contributions [4]. The K-ADAPT variant adds operators in chunks of size K, significantly reducing the total number of quantum measurements and classical optimization cycles required to achieve chemical accuracy (1.6 mHa or 1 kcal/mol).
Hamiltonian Transformation and Qubit Reduction
The fermionic Hamiltonian of quantum chemistry must be mapped to qubit operators for implementation on quantum processors. Common techniques include the Jordan-Wigner and Bravyi-Kitaev transformations [4] [6]. Additionally, quantum tapering approaches exploit molecular symmetries to reduce the number of qubits required, while quantum community detection algorithms running on quantum annealers can identify relevant Slater determinant clusters to reduce Hamiltonian complexity [3].
The following diagram illustrates the complete workflow for molecular ground state determination using quantum algorithms:
Experimental Protocols and Methodologies
VQE Implementation Protocol
A standardized protocol for VQE implementation encompasses the following steps:
Molecular Hamiltonian Preparation
- Perform Hartree-Fock calculation using quantum chemistry packages (e.g., PySCF)
- Extract one-electron (( h{pq} )) and two-electron (( h{pqrs} )) integrals
- Apply active space approximation if necessary to reduce problem size
Qubit Hamiltonian Construction
- Transform fermionic operators to qubit operators using Jordan-Wigner or Bravyi-Kitaev transformation
- Apply symmetry-based tapering to reduce qubit count
- Express Hamiltonian as a linear combination of Pauli terms: ( H = \sumi ci P_i )
Ansatz Selection and Initialization
- Choose ansatz architecture: UCCSD, hardware-efficient, or adaptive construction
- Initialize parameters using classical approximations or heuristic methods
- Prepare reference state (typically Hartree-Fock) on quantum processor
Measurement and Optimization Loop
- Measure expectation values of Hamiltonian terms
- Compute total energy: ( E(\theta) = \langle \psi(\theta) | H | \psi(\theta) \rangle )
- Update parameters using classical optimizers (COBYLA, BFGS, or CMA-ES)
- Iterate until convergence to chemical accuracy
Resource Estimation for Fault-Tolerant Algorithms
For fault-tolerant quantum computing, the quantum phase estimation algorithm requires careful resource estimation [2]. The total cost to obtain energy E with precision ε is given by:
( \text{Cost}_{\text{QPE}} = \text{poly}(1/S) \times [\text{poly}(L)\text{poly}(1/\epsilon) + C] )
where S is the initial state overlap with the true ground state, L is the system size, and C represents state preparation cost. This highlights the critical importance of high-overlap initial states for practical quantum advantage.
Computational Tools and Frameworks
Table 3: Essential Computational Tools for Molecular Ground State Research
| Tool/Resource | Type | Primary Function | Application Context |
|---|---|---|---|
| PySCF [4] | Classical quantum chemistry package | Hartree-Fock, integral computation | Hamiltonian preparation |
| OpenFermion [4] | Quantum chemistry library | Fermion-to-qubit mapping | Quantum algorithm input |
| PennyLane [6] | Quantum machine learning | Differentiable quantum computing | VQE implementation |
| QChem [6] | Software module | Molecular Hamiltonian construction | End-to-end quantum chemistry |
| D-Wave Ocean [3] | Quantum annealing SDK | QUBO problem formulation | Hamiltonian reduction |
Key Mathematical Constructs
The "Unitary Coupled Cluster with Singles and Doubles (UCCSD)" ansatz serves as a cornerstone for many quantum approaches, with the wavefunction form:
( |\Psi{\text{UCCSD}}\rangle = e^{T - T^\dagger} |\Psi{\text{HF}}\rangle )
where ( T = T1 + T2 ) with ( T1 = \sum{i,j} \theta{ij} \hat{a}i^\dagger \hat{a}j ) and ( T2 = \sum{i,j,k,l} \theta{ij}^{kl} \hat{a}i^\dagger \hat{a}j^\dagger \hat{a}k \hat{a}l ) [4].
For dissipative approaches [9], the Lindblad master equation:
( \frac{d}{dt}\rho = \mathcal{L}[\rho] = -i[\hat{H},\rho] + \sumk \hat{K}k \rho \hat{K}k^\dagger - \frac{1}{2} {\hat{K}k^\dagger \hat{K}_k, \rho} )
provides the theoretical framework for ground state preparation through engineered dissipation, with Type-I and Type-II jump operators enabling different convergence properties.
Current Challenges and Research Frontiers
The Quest for Quantum Advantage
The evidence for exponential quantum advantage in ground state quantum chemistry remains uncertain [2]. Key challenges include:
- Initial state preparation: The overlap between easily preparable states (e.g., Hartree-Fock) and the true ground state may decrease exponentially with system size due to the orthogonality catastrophe
- Classical heuristic power: Advanced classical methods (DMRG, AFQMC, neural network states) often achieve polynomial scaling for realistic chemical systems
- Error scaling: The precise relationship between system size and computational cost for fixed accuracy remains poorly characterized for both classical and quantum approaches
Emerging Approaches
Promising research directions include:
- Stabilizer ground states for initializing and analyzing quantum many-body systems [10]
- Broken-symmetry wavefunctions for improving initial state overlap in open-shell systems [7]
- Quantum-inspired classical algorithms leveraging insights from quantum information science
- Hierarchical multilevel approaches combining classical and quantum computations
The following diagram illustrates the key considerations in the quantum-classical comparison for molecular ground state problems:
The determination of molecular Hamiltonian ground states remains a vibrant research area at the intersection of quantum chemistry, computational physics, and quantum information science. While quantum computing offers promising long-term potential, particularly for strongly correlated systems that challenge classical methods, rigorous demonstration of exponential quantum advantage awaits both theoretical advancement and hardware development. The most productive near-term trajectory appears to involve co-design of quantum-classical hybrid algorithms that leverage the respective strengths of both computational paradigms, with careful attention to resource scaling, error management, and application to chemically relevant problems.
In quantum chemistry, determining the exact ground state of a molecular Hamiltonian is fundamental to predicting a molecule's properties and behavior. For systems where electrons are strongly correlated—such as transition metal complexes, catalysts, and high-temperature superconductors—this task becomes profoundly difficult. Classical computational methods face an exponential wall, a problem where the computational resources required to find an exact solution grow exponentially with the size of the system [11]. This scaling relationship poses a fundamental limit to what is computationally tractable, even on the most powerful classical supercomputers.
This article explores the nature of this exponential wall, details the limitations of current classical algorithms, and examines how quantum computing offers a promising pathway to overcome these barriers, ultimately enabling the accurate simulation of complex molecular systems that are critical for advancements in drug discovery and materials science [12] [13].
The Exponential Wall in Classical Computational Chemistry
The Source of the Exponential Scaling
The exponential wall arises from the fundamental nature of quantum mechanics. A multi-electron wavefunction, which describes the state of a system, can be represented as a linear combination of Slater determinants. Each determinant represents a possible electronic configuration. For an exact solution—a method known as Full Configuration Interaction (FCI)—one must consider all possible excitations of electrons across the available orbitals [11].
The number of these determinants does not grow linearly or polynomially with system size; it scales combinatorially. For even modestly sized molecules (e.g., more than four heavy atoms) and basis sets, the number of determinants becomes astronomically large. Consequently, solving for the coefficients in the wavefunction expansion requires diagonalizing a Hamiltonian matrix whose dimension is this vast number of determinants, an operation whose cost scales as the cube of the matrix dimension. This combinatorial explosion of problem size, combined with the polynomial cost of linear algebra, creates the intractable "exponential wall" [11].
Limitations of Approximate Classical Methods
To make simulations feasible, quantum chemists have developed approximate methods that avoid the full FCI solution. Table 1 summarizes the key classical approaches and their limitations when dealing with strongly correlated systems.
Table 1: Classical Computational Methods for Ground-State Quantum Chemistry
| Method | Key Principle | Scaling with System Size | Limitations with Strong Correlation |
|---|---|---|---|
| Density Functional Theory (DFT) | Uses electron density instead of wavefunction; relies on approximate functionals. | Polynomial [13] | Standard functionals fail for strongly correlated electrons; inaccurate for bond breaking, transition metals, and catalytic sites [12] [13]. |
| Coupled Cluster (CC) | Uses an exponential ansatz of excitation operators. | High-order polynomial (e.g., CCSD(T) scales as ~N⁷) | Fails when the reference wavefunction (often from Hartree-Fock) is a poor starting point, as in multi-reference systems [11]. |
| Complete Active Space SCF (CASSCF) | Performs FCI within a selected "active space" of important orbitals. | Exponentially scaling with active space size [11] | The exponential wall reappears as the active space grows to capture more correlation; tractable only for small active spaces. |
| Density Matrix Renormalization Group (DMRG) | Matrix Product State ansatz; iterative variational optimization. | Polynomial for weakly correlated 1D systems [11] | For strongly correlated systems, the scaling becomes unfavorable, and the computational cost increases dramatically [11]. |
As the table indicates, each classical method involves a trade-off between computational cost and accuracy. Strongly correlated systems, which are central to many important chemical problems like nitrogen fixation [13] and enzymatic catalysis [14], often fall into the gap where approximations fail and exact methods are too costly.
Quantum Computing: A Path Beyond the Exponential Wall
The Quantum Approach to Electronic Structure
Quantum computers naturally circumvent the exponential wall by leveraging the principles of superposition and entanglement. While a classical computer must store and process each of the exponentially many configurations of a quantum state separately, a quantum computer can encode this information in the amplitudes of a quantum state of just n qubits, which can represent 2ⁿ states simultaneously [12] [13].
The Quantum Phase Estimation (QPE) algorithm can, in principle, project onto the exact ground state of a Hamiltonian and determine its energy with high precision. For the Noisy Intermediate-Scale Quantum (NISQ) era, hybrid classical-quantum algorithms like the Variational Quantum Eigensolver (VQE) have been developed. In VQE, a quantum processor prepares and measures a parameterized trial wavefunction (ansatz), while a classical optimizer adjusts the parameters to minimize the expectation value of the energy [12]. This approach trades circuit depth for increased measurements and is more resilient to noise.
Documented Quantum Speedups and Current Capabilities
While fault-tolerant quantum computers capable of solving large chemistry problems are still under development, progress is accelerating. Recent industry roadmaps project systems with 1,000+ physical qubits in the near term, with plans for error-corrected systems featuring hundreds of logical qubits by the end of the decade [15]. Table 2 quantifies the requirements and recent demonstrations of quantum chemistry simulations.
Table 2: Quantum Computing for Molecular Hamiltonians: Requirements and Demonstrations
| Metric / Molecule | Classical FCI Requirement | Quantum Computing Demonstration / Requirement | Key Finding / Implication |
|---|---|---|---|
| General Qubit Requirement | N/A | Problem encoded onto ~Polynomial(n) qubits [12] | Exponential compression of the problem representation. |
| FeMoco (Nitrogenase) | Intractable for FCI [13] | Estimated at ~2.7 million physical qubits (pre-error correction) [13] | Highlighed the qubit count challenge; recent advances in error correction and algorithms are reducing this figure. |
| Cytochrome P450 | Intractable for FCI [13] | Similar scale to FeMoco [13] | A key industrial target for drug metabolism simulation. |
| Small Molecules (H₂, LiH) | Tractable, but limited | VQE demonstrated on NISQ devices [13] | Proof-of-principle for hybrid quantum-classical algorithms. |
| Water Molecule | Tractable | Ground state simulated via adiabatic state preparation [16] | Demonstrates advanced state preparation methods on quantum simulators. |
| Quantum Error Correction | N/A | Error rates reduced to ~0.000015%; algorithmic fault tolerance reduces overhead 100x [15] | Critical for making large-scale quantum chemistry simulations feasible. |
These developments indicate that quantum computing is steadily progressing toward solving chemically relevant problems. For instance, Google's Quantum Echoes algorithm has demonstrated a verifiable quantum advantage by running an algorithm 13,000 times faster than classical supercomputers, and collaborations between companies like IonQ and Ansys have shown quantum simulations outperforming classical high-performance computing by 12% in a medical device simulation [15].
Experimental Protocols for Quantum Ground-State Simulation
This section outlines two primary methodological frameworks for finding molecular ground states on quantum computers.
The Variational Quantum Eigensolver (VQE) Protocol
The VQE is a hybrid protocol designed for today's noisy quantum processors [13]. The workflow, as shown in Diagram 1, involves iterative communication between a quantum and a classical computer.
Diagram 1: Variational Quantum Eigensolver (VQE) Workflow
Key Steps in the VQE Protocol:
- Problem Mapping: The second-quantized molecular electronic Hamiltonian is encoded into qubits using a transformation such as the Jordan-Wigner or Bravyi-Kitaev transformation [12].
- Ansatz Preparation: A parameterized quantum circuit (ansatz) is chosen. Common choices include the Unitary Coupled Cluster (UCC) ansatz or hardware-efficient ansatzes.
- Quantum Execution: The quantum processor prepares the state
|ψ(θ)⟩and performs measurements to estimate the expectation value of the Hamiltonian,⟨ψ(θ)|H|ψ(θ)⟩. This often requires measuring the expectation values of each Pauli term in the qubit Hamiltonian. - Classical Optimization: A classical optimizer (e.g., gradient descent) uses the energy reported from the quantum computer to update the parameters
θfor the next iteration. This loop continues until the energy converges to a minimum.
Adiabatic State Preparation (ASP) Protocol
ASP is an alternative method that leverages the quantum adiabatic theorem. The protocol, detailed in Diagram 2, involves evolving the system from a simple, easy-to-prepare ground state to the complex ground state of the target molecular Hamiltonian [16].
Diagram 2: Adiabatic State Preparation (ASP) Workflow
Key Steps in the ASP Protocol:
- Define Hamiltonians: Identify an initial Hamiltonian
H₀whose ground state is known and easy to prepare on the quantum computer (e.g., the ground state of a non-interacting system). Define the target molecular HamiltonianHₜ. - Prepare Initial State: Initialize the quantum computer into the ground state of
H₀. - Adiabatic Evolution: Slowly evolve the system's Hamiltonian from
H₀toHₜover a total timeT, typically following a pathH(s) = (1-s)H₀ + sHₜ, wheresgoes from 0 to 1. The evolution must be slow enough to satisfy the adiabatic condition and prevent transitions to excited states. - Final Measurement: After the evolution, the system will reside in the ground state of
Hₜ, and its properties can be measured directly.
A recent advancement, as demonstrated for molecules like water and methylene, involves constructing an optimal adiabatic path starting from an approximate initial wavefunction (e.g., from a classical computation), rather than a trivial H₀ [16]. This heuristic method can significantly improve the efficiency of ASP.
Table 3 catalogs key algorithmic and hardware "reagent solutions" essential for conducting ground-state searches for molecular Hamiltonians.
Table 3: Essential "Research Reagents" for Quantum Computational Chemistry
| Category / Item | Function | Application in Ground-State Search |
|---|---|---|
| Algorithmic Reagents | ||
| VQE (Variational Quantum Eigensolver) | Hybrid quantum-classical algorithm for finding ground states on NISQ devices. | Robust to noise; used to simulate small molecules like H₂ and LiH [13]. |
| QPE (Quantum Phase Estimation) | Coherent algorithm for high-precision energy measurement. | Provides exact energy; requires fault-tolerant hardware [12]. |
| Quantum Krylov Methods | Projects the Hamiltonian into a subspace generated by real-time evolution. | An alternative to VQE for obtaining ground and excited states [12]. |
| Hardware Reagents | ||
| Superconducting Qubits | Quantum processors based on superconducting circuits. | Used by Google, IBM; dominant platform for current experiments [15]. |
| Trapped Ions | Quantum processors using trapped atomic ions as qubits. | Used by IonQ; known for high fidelity and long coherence [12] [15]. |
| Neutral Atoms | Quantum processors using neutral atoms in optical tweezers. | Platform used by companies like Atom Computing; offers scalability potential [15]. |
| Enabling Software | ||
| Jordan-Wigner / Bravyi-Kitaev | Fermion-to-Qubit Mapping transforms electronic Hamiltonians into qubit operators. | Encodes the molecular Hamiltonian of Fermionic systems into a form a quantum computer can process [12]. |
| UCC Ansatz | A chemically inspired ansatz for the VQE algorithm. | Generates trial wavefunctions based on electronic excitations [13]. |
The exponential wall presents a fundamental barrier to the classical simulation of strongly correlated molecular systems, hindering progress in drug discovery and materials science. While classical methods provide approximate solutions, they often fail for critical industrial problems like modeling catalytic active sites and metalloenzymes. Quantum computing, by its very nature, offers a pathway to scale polynomially rather than exponentially with system size. Although significant challenges in qubit count and error correction remain, rapid hardware advancements and algorithmic innovations are steadily transforming this promise into a tangible reality. The transition from simulating simple diatomic molecules to complex biomolecules is underway, marking the beginning of a new era for computational quantum chemistry.
The search for the ground state energy of molecular Hamiltonians represents a cornerstone problem in computational chemistry and drug discovery, with direct applications in predicting molecular reactivity, stability, and properties. Classical computational methods, particularly for strongly correlated molecules, require expensive wave function methods that become prohibitively costly even for few-atom systems [17]. Quantum computing emerges as a transformative solution to this enduring challenge by harnessing the distinct properties of quantum mechanics—superposition and entanglement—to process information in ways fundamentally inaccessible to classical computers.
This technical guide examines the pathway from classical bits to quantum bits (qubits), focusing on how these quantum properties enable computational advantage in molecular ground state searches. Unlike classical bits that are confined to definite states of 0 or 1, qubits can exist in a coherent superposition of both states simultaneously [18] [19]. Furthermore, through entanglement, qubits become intrinsically linked, creating correlations that persist even when physically separated [20]. When combined, these phenomena enable a quantum computer to represent and manipulate molecular wavefunctions in an exponentially large computational space, offering a potentially powerful alternative to classical computational chemistry methods for tackling the electronic structure problem [21] [22].
Theoretical Foundations: From Bits to Qubits
The Classical Bit: A Binary Foundation
The classical bit is the fundamental unit of information in traditional computing. It is a binary system that can exist in only one of two mutually exclusive states, physically represented by a high or low voltage in a circuit, and logically interpreted as 0 or 1 [19]. This deterministic nature means that a system of n classical bits can be in exactly one of 2^n possible configurations at any given time. While this paradigm has powered decades of computational advancement, its sequential processing nature creates a fundamental bottleneck for simulating quantum mechanical systems, which do not abide by these classical constraints.
The Quantum Bit: A Superposition of Possibilities
The qubit, the quantum analog of the classical bit, leverages the principles of quantum mechanics to transcend binary limitations. Like a classical bit, a qubit can be measured in the basis states |0⟩ or |1⟩. However, before measurement, it can exist in any coherent superposition of these states, represented as |ψ⟩ = α|0⟩ + β|1⟩, where α and β are complex probability amplitudes such that |α|^2 + |β|^2 = 1 [18] [22]. Upon measurement, the qubit's state collapses to either |0⟩ or |1⟩, with probabilities |α|^2 and |β|^2 respectively.
The power of quantum computing scales exponentially with the number of qubits. While n classical bits can represent only one of 2^n states at a time, n qubits can exist in a superposition of all 2^n possible states simultaneously [20]. This exponential scaling provides the theoretical foundation for quantum advantage, allowing quantum algorithms to explore a vast solution space in parallel.
Quantum Entanglement: Generating Non-Classical Correlations
Entanglement is a uniquely quantum phenomenon where the quantum states of two or more qubits become inextricably linked, such that the state of one cannot be described independently of the others [18]. Measuring one entangled qubit instantaneously affects the state of its partner, regardless of the physical distance between them [20]. This "spooky action at a distance," as Einstein termed it, creates powerful non-classical correlations that are essential for quantum computation and communication. Entanglement enables operations on one qubit to have a cascading effect on many others, allowing for a high degree of coordination and information encoding that is impossible for classical systems [20].
Table 1: Fundamental Properties of Classical Bits vs. Quantum Bits
| Property | Classical Bit | Quantum Bit (Qubit) | ||||
|---|---|---|---|---|---|---|
| State | Definitively 0 or 1 | Superposition of | 0⟩ and | 1⟩ (`α | 0⟩ + β | 1⟩`) |
| Information Capacity | 1 bit per bit | Theoretically infinite (before measurement) | ||||
System of n Units |
Can represent one of 2^n states |
Can represent a superposition of all 2^n states |
||||
| Inter-Particle Correlation | Independent | Can be entangled, creating non-classical correlations | ||||
| Governing Physics | Classical Mechanics | Quantum Mechanics |
The Molecular Hamiltonian and the Ground State Problem
Defining the Electronic Hamiltonian
In computational chemistry, the goal is to solve the electronic Schrödinger equation to determine a molecule's properties. Within the Born-Oppenheimer approximation, which treats nuclei as fixed point charges, the electronic Hamiltonian H_e captures the energy contributions from electron kinetic energy and all Coulomb interactions (electron-nucleus attraction, electron-electron repulsion, and nucleus-nucleus repulsion) [21]. The total electronic energy E and the wave function Ψ are obtained by solving H_e Ψ = E Ψ. The lowest eigenvalue E_g is the ground state energy, and its corresponding eigenvector Ψ_g is the ground state wave function.
The Challenge of Strong Correlation
Classical algorithms for predicting the equilibrium geometry of strongly correlated molecules require expensive wave function methods that become impractical already for few-atom systems [17]. The computational cost of exact methods scales exponentially with the number of electrons, making them intractable for all but the smallest molecules. While approximation methods exist, they often trade accuracy for feasibility, particularly for systems where electron correlation is strong, such as in transition metal complexes or reaction transition states, which are critical in pharmaceutical research.
Second Quantization and Qubit Mapping
To simulate molecules on a quantum computer, the electronic Hamiltonian must be translated into a form that operates on qubits. This is typically done using the second quantization formalism. In this representation, the Hamiltonian H is expressed in terms of creation (c_p^†) and annihilation (c_q) operators as [21]:
The coefficients h_{pq} and h_{pqrs are one- and two-electron integrals computed using a chosen basis set of molecular orbitals.
The next step is to map these fermionic operators to qubit operators. This is achieved using techniques like the Jordan-Wigner or Bravyi-Kitaev transformations, which encode the occupation number of each molecular orbital into the state of a qubit [21]. These transformations express the Hamiltonian as a linear combination of tensor products of Pauli operators (I, X, Y, Z):
This Pauli string representation is executable on a quantum processor.
Leveraging Quantum Properties for Ground State Search
The Role of Superposition in State Preparation
Superposition is instrumental in preparing an initial ansatz for the molecular wavefunction. A quantum computer can efficiently generate a trial state that is a superposition of many different electronic configurations simultaneously. For instance, starting all qubits in the |0⟩ state and applying Hadamard gates creates a uniform superposition of all possible computational basis states. This state can then be evolved or parameterized to approach the true, complex ground state of the target molecular Hamiltonian. This ability to compactly represent a vast set of configurations is a direct advantage over classical computers, which must often enumerate configurations individually.
The Role of Entanglement in Capturing Electron Correlation
Entanglement is the quantum resource that directly encodes electron correlations. In a molecular system, the motion of electrons is correlated; the position of one electron provides information about the likely positions of others. Classical methods struggle to represent these complex, multi-electron correlations efficiently. On a quantum computer, entangling gates (such as CNOT gates) applied between qubits create these necessary correlations in the wavefunction ansatz [23]. The degree and structure of entanglement available in a variational quantum algorithm significantly impact its convergence and ability to accurately approximate the ground state, particularly for strongly correlated systems [23].
Interference and Amplitude Amplification
While not a core pillar like superposition and entanglement, quantum interference is the process that amplifies correct answers and suppresses incorrect ones. After preparing a superposed and entangled state, a quantum algorithm manipulates the probability amplitudes (α, β) of different states such that those corresponding to the ground state energy constructively interfere, while others destructively interfere. This process effectively "rotates" the quantum state in the high-dimensional Hilbert space toward the desired solution. When the system is finally measured, the probability of obtaining an outcome corresponding to the ground state energy is maximized.
Algorithmic Frameworks and Experimental Protocols
The Variational Quantum Eigensolver (VQE) Protocol
The VQE algorithm is a leading hybrid quantum-classical approach for ground state finding in the NISQ era. It combines a quantum computer's ability to prepare and measure complex states with a classical computer's power to optimize parameters.
Detailed Methodology:
- Problem Specification: Define the target molecule (atomic symbols and nuclear coordinates) and a basis set [21].
- Hamiltonian Generation: Use a classical computer to compute the one- and two-electron integrals (
h_{pq},h_{pqrs}) and transform the electronic Hamiltonian into a qubit-representable form (a sum of Pauli strings) [21]. - Ansatz Preparation: On the quantum computer, prepare a parameterized trial wavefunction (ansatz)
|ψ(θ)⟩ = U(θ)|ψ_0⟩, whereU(θ)is a parameterized quantum circuit, and|ψ_0⟩is a simple reference state (e.g., the Hartree-Fock state) [17]. - Expectation Value Measurement: For each Pauli term
P_iin the HamiltonianH = Σ_i c_i P_i, measure the expectation value⟨ψ(θ)|P_i|ψ(θ)⟩on the quantum processor. The total energy estimate isE(θ) = Σ_i c_i ⟨ψ(θ)|P_i|ψ(θ)⟩. - Classical Optimization: A classical optimizer (e.g., gradient descent) uses the energy
E(θ)to propose new parametersθ_newto minimize the energy. - Iteration: Steps 3-5 are repeated until convergence, at which point
E(θ*)is the estimated ground state energy.
Diagram Title: VQE Workflow for Molecular Ground State Search
Advanced Protocol: Qubit Configuration Optimization for Neutral Atoms
For specific hardware platforms like neutral atom tweezers, where qubit interactions are determined by their physical positions, advanced configuration optimization can dramatically improve VQE performance.
Detailed Methodology (Consensus-Based Optimization) [23]:
- Initialization: Multiple "agents" (each a set of qubit positions
X^(k)) are randomly initialized in the configuration space𝒳. - Partial Pulse Optimization: For each agent's configuration
X^(k), a partial optimization of the control pulsesz^(k)is performed to get an initial indication of the cost landscapeJ(X, z). - Consensus Update: The agents' cost function values and positions are shared. A weighted consensus point is computed, favoring agents with lower costs.
- Exploration and Diffusion: Agents are updated by moving toward the consensus point while adding noise for exploration and a diffusion term to avoid premature convergence.
- Iteration and Selection: Steps 2-4 are repeated for several iterations. The positions converge to an optimized configuration, which is then used for a full VQE run. This method avoids the pitfalls of gradient-based position optimization, which fails due to the divergent
R^(-6)nature of Rydberg interactions [23].
Diagram Title: CBO for Qubit Configuration Optimization
Quantitative Performance and Resource Analysis
The practical utility of quantum algorithms depends on their performance relative to resource constraints, particularly in the NISQ era. Recent research demonstrates significant progress in making Hamiltonian simulation more feasible.
Table 2: Algorithmic Performance and Resource Reduction in Hamiltonian Simulation
| Metric | Classical / Baseline Method | Quantum-Enhanced Approach | Improvement Factor |
|---|---|---|---|
| Circuit Depth (All-to-All connectivity) | Baseline | Hamiltonian truncation + Clifford Decomposition (CDAT) [24] | 28.5-fold reduction |
| Circuit Depth (IBMQ Heron) | Baseline | Hamiltonian truncation + Clifford Decomposition (CDAT) [24] | 15.5-fold reduction |
| Convergence Acceleration | Standard fixed configuration VQE | VQE with Consensus-Based qubit configuration optimization [23] | Significant acceleration and lower error |
| Mitigation of Barren Plateaus | Standard ansatz initialization | Problem-inspired ansatz & optimized qubit positions [23] | Helps avoid flat, untrainable regions |
A 2024 study on simulating pharmaceutically relevant molecules with sulfonyl fluoride warheads achieved a reduction in circuit depth to 1330 gates for an 8-qubit Hamiltonian. Using middleware decomposition, they successfully executed sub-circuits with depths of 371 gates (containing 216 2-qubit gates) on current hardware, representing one of the largest electronic structure dynamics calculations implemented to date [24].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Quantum Computational Chemistry
| Resource / "Reagent" | Type | Function / Application | Example Platforms / Libraries |
|---|---|---|---|
| Superconducting Qubits | Hardware | Fast, controllable physical qubits; common in commercial processors. | IBM Condor, Google Willow |
| Trapped Ion Qubits | Hardware | High-fidelity qubits with long coherence times; well-suited for quantum simulations. | IonQ |
| Neutral Atom Arrays | Hardware | Configurable qubit interactions; enables position optimization for specific problems [23]. | Cold atom platforms |
| PennyLane | Software | A cross-platform Python library for differentiable programming of quantum computers; facilitates quantum chemistry simulations [21]. | Xanadu |
| Qiskit | Software | An open-source SDK for working with quantum computers at the level of circuits, pulses, and algorithms [22]. | IBM |
| Molecular Hamiltonian | Input Data | The target operator encoding the molecular energy; the core object of the simulation. | Generated via qchem.molecular_hamiltonian() [21] |
| Variational Quantum Eigensolver (VQE) | Algorithm | A hybrid algorithm for finding ground states, robust to NISQ-era noise. | Standard in QC libraries |
| Consensus-Based Optimization (CBO) | Algorithm | A gradient-free method for optimizing qubit configurations in neutral atom systems [23]. | Custom implementation per research |
| Problem-Inspired Ansatz | Algorithm | An ansatz (e.g., UCCSD, ADAPT-VQE) tailored to the problem, reducing circuit depth vs. universal ansatzes [23]. | Common in quantum chemistry |
The transition from bits to qubits, powered by the fundamental principles of superposition and entanglement, marks a paradigm shift in computational science with profound implications for molecular research. By providing a natural framework for representing and manipulating complex quantum systems, quantum computing offers a viable path to solving the electronic structure problem for molecules that are currently beyond the reach of classical computers. While significant challenges remain—including qubit decoherence, noise, and the need for robust error correction [25] [19]—the methodological advances in variational algorithms, qubit configuration optimization, and circuit depth reduction are steadily paving the way for quantum utility in drug development and materials discovery. The ongoing symbiosis of algorithmic innovation and hardware progress promises to unlock new frontiers in understanding and designing molecular systems.
The quest for quantum utility—the point at which quantum computers solve practically valuable problems beyond the reach of classical systems—represents a pivotal challenge in computational science. For researchers focused on molecular Hamiltonian ground state searches, understanding the fundamental relationships between quantum and classical complexity classes is essential for navigating this rapidly evolving landscape. The complexity classes BQP (Bounded-error Quantum Polynomial time) and BPP (Bounded-error Probabilistic Polynomial time) serve as critical landmarks in this exploration, defining the boundaries of efficient computation on quantum and classical devices, respectively [26] [27].
Within computational chemistry, the ground state energy estimation problem provides a compelling case study for examining these complexity relationships. While classical methods like Density Matrix Renormalization Group (DMRG) and tensor networks have demonstrated considerable success, they face exponential scaling walls for strongly correlated systems prevalent in catalytic and biomolecular applications [28] [12]. Quantum computing offers a potential pathway through polynomial-time solutions to these otherwise intractable problems, positioning ground state estimation as a problem likely residing in BQP but not in BPP [12] [29].
This technical analysis examines the current understanding of the BQP-BPP relationship through the lens of molecular Hamiltonian problems, surveys established and emerging quantum algorithmic approaches, and presents recent experimental evidence suggesting the imminent realization of quantum utility in computational chemistry.
The Computational Complexity Landscape
Defining the Complexity Classes
The complexity classes BPP and BQP define the sets of decision problems solvable by probabilistic classical and quantum computers, respectively, with bounded error probability in polynomial time [26] [27].
- BPP: A decision problem is in BPP if a probabilistic Turing machine can solve it in polynomial time with an error probability of at most 1/3 for all instances. The error bound can be made exponentially small through repetition [26].
- BQP: Similarly, BQP contains decision problems solvable by a quantum computer in polynomial time with error probability at most 1/3. The quantum circuit model serves as the formal model of computation for BQP [27].
The table below summarizes the fundamental relationships between key complexity classes relevant to quantum chemistry applications:
Table 1: Computational Complexity Class Relationships
| Complexity Class | Computational Model | Error Bound | Inclusion Relationships |
|---|---|---|---|
| P | Deterministic Turing machine | None | P ⊆ BPP [26] |
| BPP | Probabilistic Turing machine | ≤ 1/3 | BPP ⊆ BQP [26] [27] |
| BQP | Quantum computer | ≤ 1/3 | BQP ⊆ PP ⊆ PSPACE [26] [27] |
| NP | Non-deterministic Turing machine | None | BQP relationship with NP unknown [27] |
| PSPACE | Deterministic Turing machine with polynomial space | None | BQP ⊆ PSPACE [27] |
The Fundamental Question: BQP vs. BPP
The central open question in quantum complexity theory relevant to quantum utility is whether BPP ⊊ BQP—that is, whether quantum computers can solve problems that classical computers cannot efficiently solve [26] [27]. While it is known that BPP ⊆ BQP, meaning all efficiently solvable classical problems are also efficiently solvable on quantum computers, the converse containment remains unproven [26].
For practical quantum chemistry applications, this theoretical question translates to whether quantum algorithms can provide superpolynomial speedups for real molecular systems. The existence of such speedups remains actively debated, with evidence suggesting that quantum advantage may be most pronounced for dynamics simulations and strongly correlated systems [12].
Quantum Algorithms for Molecular Ground State Problems
Algorithmic Approaches and Their Complexities
Multiple quantum algorithmic frameworks have been developed for ground state energy estimation, each with distinct resource requirements and complexity profiles:
Table 2: Quantum Algorithms for Ground State Energy Estimation
| Algorithm | Key Principle | Theoretical Complexity | Fault-Tolerance Requirement | Application Scale Demonstrated |
|---|---|---|---|---|
| Variational Quantum Eigensolver (VQE) [12] | Hybrid quantum-classical optimization | Heuristic; depends on ansatz | No (NISQ-compatible) | 12+ qubits [28] |
| Quantum Phase Estimation (QPE) [29] | Quantum Fourier transform of phase | O(1/ϵ) depth for precision ϵ | Yes | Small-scale fault-tolerant simulations |
| Quantum Selected CI (QSCI) [28] | Quantum-assisted configuration interaction | Beyond classical exact diagonalization | Partial (with error mitigation) | Up to 77 qubits [28] |
| CDF-Based Methods [29] | Cumulative distribution function analysis | O(polylog(n)) for sparse systems | Early fault-tolerant | 26-qubit simulations [29] |
The Role of Hybrid Quantum-Classical Approaches
Hybrid quantum-classical algorithms represent a pragmatic approach to leveraging current quantum hardware within the broader context of computational chemistry workflows. The quantum-selected configuration interaction (QSCI) method demonstrates how quantum processors can be deployed for specific, computationally challenging subroutines within larger classical computational frameworks [28]. This approach aligns with the increasingly common integration of Quantum Processing Units (QPUs) with conventional High-Performance Computing (HPC) platforms, creating heterogeneous systems capable of addressing complex multiscale simulations [28].
For molecular systems, these hybrid approaches often employ embedding techniques such as projection-based embedding (PBE) and density matrix embedding theory (DMET) to partition the computational problem into quantum and classical tractable subdomains [28]. This strategic partitioning enables the application of quantum resources to the most electronically correlated regions of large molecular systems while treating the broader environmental effects through classical molecular mechanics or density functional theory.
Experimental Evidence: Toward Quantum Utility
Recent Experimental Demonstrations
Recent experiments on noisy intermediate-scale quantum (NISQ) devices have provided preliminary evidence for the utility of quantum computing before full fault tolerance. A landmark 2023 study on a 127-qubit superconducting processor demonstrated the measurement of accurate expectation values for circuit volumes beyond brute-force classical computation [30]. This experiment implemented Trotterized time evolution of a 2D transverse-field Ising model, with results in the strong entanglement regime where classical tensor network methods break down [30].
Crucially, these experiments leveraged advanced error mitigation techniques, including zero-noise extrapolation (ZNE) and probabilistic error cancellation (PEC), to extract accurate expectation values from noisy quantum circuits [30]. The successful application of these techniques to circuits with up to 2,880 CNOT gates suggests a viable path toward quantum utility for chemical applications in the pre-fault-tolerant era.
Experimental Protocol: Quantum Utility Demonstration
The experimental methodology for demonstrating quantum utility in chemical systems typically follows this structured protocol:
System Selection: Identify a molecular system with strong electron correlation effects that challenge classical methods (e.g., multimetallic catalysts or conjugated polymers) [28] [12].
Problem Decomposition: Apply embedding techniques to partition the system into active quantum and environmental classical regions using methods like QM/MM or DMET [28].
Algorithm Implementation: Execute hybrid quantum-classical algorithms (VQE, QSCI) on integrated HPC-QPU platforms, employing qubit subspace techniques to reduce quantum resource requirements [28].
Error Mitigation: Apply advanced error mitigation protocols (ZNE, PEC) to extract accurate expectation values from noisy quantum computations [30].
Classical Verification: Compare results with exactly solvable test cases and state-of-the-art classical methods to verify accuracy where possible [30].
The workflow diagram below illustrates this multi-scale computational approach:
The Scientist's Toolkit: Research Reagent Solutions
For researchers implementing quantum computational methods for molecular Hamiltonian problems, the following "research reagents" represent essential components of the experimental framework:
Table 3: Essential Research Reagents for Quantum Computational Chemistry
| Research Reagent | Function | Implementation Examples |
|---|---|---|
| Error Mitigation Protocols [30] | Reduce noise impact in NISQ era | Zero-noise extrapolation (ZNE), Probabilistic error cancellation (PEC) |
| Embedding Techniques [28] | Partition large systems into quantum/classical regions | Projection-based embedding (PBE), Density matrix embedding theory (DMET) |
| Qubit Subspace Methods [28] | Reduce quantum resource requirements | Qubit tapering, Contextual subspace VQE |
| Classical-Quantum Hybridizers [28] [29] | Interface quantum and classical resources | QM/MM frameworks, HPC-QPU integration platforms |
| State Preparation Protocols [29] | Initialize quantum states for computation | ADAPT-VQE, Quantum Krylov methods, DMRG-inspired initial states |
The path to demonstrable quantum utility in molecular Hamiltonian ground state calculations requires continued progress along multiple fronts: improvement in quantum hardware coherence and scalability, development of more efficient quantum algorithms with proven speedups, and refinement of error mitigation techniques that bridge the NISQ and fault-tolerant eras. The current evidence suggests that hybrid quantum-classical approaches employing strategic embedding and resource reduction techniques offer the most viable near-term pathway [28].
For computational chemists and drug development professionals, the practical implication is that quantum computational methods are transitioning from theoretical constructs to potentially valuable tools for specific, classically challenging problems in molecular systems. The complexity landscape defined by BQP and BPP provides both a target for algorithmic development and a framework for assessing progress toward genuine quantum utility in real-world chemical applications.
From Algorithms to Action: Practical Methods for Ground State Search on Quantum Hardware
The accurate calculation of ground state energies of molecular systems is a cornerstone problem in quantum chemistry and drug discovery. This challenge, however, is classically intractable for large systems due to the exponential scaling of the Hilbert space. Quantum computing offers a promising path forward, with three primary algorithmic paradigms emerging for the molecular ground state search: the Variational Quantum Eigensolver (VQE), Quantum Krylov Subspace Methods, and Quantum Phase Estimation (QPE). This whitepaper provides an in-depth technical analysis of these core algorithms, comparing their theoretical foundations, resource requirements, and practical implementation for molecular Hamiltonian problems relevant to pharmaceutical research.
Algorithmic Foundations & Comparative Analysis
Variational Quantum Eigensolver (VQE)
VQE is a hybrid quantum-classical algorithm that applies the variational principle to find the ground state energy of a Hamiltonian [31] [32]. The algorithm prepares a parameterized trial state (ansatz) |ψ(θ⃗)⟩ on a quantum computer and measures the expectation value ⟨H⟩ = ⟨ψ(θ⃗)|H|ψ(θ⃗)⟩. A classical optimizer then adjusts the parameters θ⃗ to minimize this expectation value [33].
The molecular Hamiltonian H must first be mapped to qubit operators, typically via the Jordan-Wigner or Bravyi-Kitaev transformation, expressing H as a sum of Pauli strings: H = Σᵢ αᵢPᵢ, where Pᵢ are tensor products of Pauli operators [32]. Each term's expectation value is measured separately, and the results are combined classically. Common ansätze include the Unitary Coupled Cluster (UCC) and hardware-efficient ansätze [33] [34].
Quantum Krylov Subspace Methods
Quantum Krylov algorithms diagonalize the Hamiltonian within a subspace spanned by quantum states {|ψ⟩, U|ψ⟩, U²|ψ⟩, ..., U^(k-1)|ψ⟩}, where U is typically the time-evolution operator e^(-iHt) [35] [36]. The ground state energy is estimated by solving a generalized eigenvalue problem in this subspace: Hc = ESc, where Hᵢⱼ = ⟨ψᵢ|H|ψⱼ⟩ and Sᵢⱼ = ⟨ψᵢ|ψⱼ⟩ are measured on the quantum computer [36].
Recent advances like Mirror Subspace Diagonalization (MSD) address the significant sampling cost challenge by expressing the Hamiltonian as a linear combination of time-evolution unitaries with symmetrically shifted timesteps, approaching the theoretical lower bound of sampling cost for quantum Krylov algorithms [35]. These methods are particularly effective when the spectral norm of the Hamiltonian is significantly smaller than its 1-norm [35].
Quantum Phase Estimation (QPE)
QPE is a fundamental quantum algorithm that estimates the phase ϕ of an eigenvalue e^(iϕ) of a unitary operator U, where U|ψ⟩ = e^(iϕ)|ψ⟩ for an eigenstate |ψ⟩ [37]. For Hamiltonian simulation, U is typically chosen as the time-evolution operator e^(-iHt), and the phase ϕ is related to the energy E by ϕ = Et [37].
The algorithm uses two registers: an estimation register with n qubits and a state register initially containing |ψ⟩. After applying Hadamard gates to the estimation register, controlled-U^(2^k) operations are applied for k = 0 to n-1, followed by an inverse Quantum Fourier Transform to extract the phase estimate [37] [38]. The precision of the estimate scales with the number of estimation qubits, with an error that decreases exponentially at the cost of exponentially increasing circuit depth [37].
Filtered Quantum Phase Estimation (FQPE) has recently been developed to mitigate the unfavorable dependence on initial state overlap present in standard QPE [39]. This approach uses spectral filtering to enhance the overlap of the input state with the desired eigenstate, with numerical experiments on Fermi-Hubbard models showing runtime reductions of more than two orders of magnitude in the high-precision regime [39].
Comparative Performance Analysis
Table 1: Comparative Analysis of Quantum Ground State Algorithms
| Algorithm | Theoretical Foundation | Circuit Depth | Sampling Complexity | Classical Co-processing | Current Hardware Suitability | ||
|---|---|---|---|---|---|---|---|
| VQE | Variational principle [31] [32] | Low (NISQ-friendly) | O( | γ₀ | ⁻⁴ ΔE₀ ε⁻²) for QCELS variant [39] | Extensive (parameter optimization) | High (runs on NISQ devices) [32] |
| Quantum Krylov | Subspace diagonalization [36] | Moderate | Near-optimal up to logarithmic factor [35] | Moderate (subspace eigenvalue problem) | Medium (early fault-tolerant) | ||
| QPE | Quantum Fourier transform [37] | High (exponential in precision) | O~(ε⁻¹ + | γ₀ | ⁻²ΔE₀⁻¹) for FQPE variant [39] | Minimal (classical post-processing) | Low (requires fault-tolerance) [38] |
Table 2: Algorithmic Resource Requirements and Performance Characteristics
| Algorithm | Initial Overlap Dependence | Convergence Guarantees | Measurement Cost | Key Limitations | |
|---|---|---|---|---|---|
| VQE | Moderate | No guarantee (barren plateaus) [32] | High (many Pauli terms) | Ansatz design, optimization challenges [32] | |
| Quantum Krylov | Moderate to high | Provable convergence under conditions [36] | Medium to high (matrix elements) | Ill-conditioned overlap matrices [36] | |
| Standard QPE | Strong (success probability ∝ | γ₀ | ²) [39] | Deterministic with perfect circuits | Exponential circuit depth [38] |
| FQPE | Reduced via filtering [39] | Deterministic with perfect circuits | Similar to QPE with improved success | Filter implementation complexity |
Experimental Protocols & Methodologies
VQE Implementation for H₂ Molecule
The VQE protocol for the hydrogen molecule demonstrates the standard experimental workflow [33]:
Hamiltonian Preparation: The molecular Hamiltonian for H₂ is constructed in the STO-3G basis set using the Jordan-Wigner transformation, resulting in a 4-qubit Hamiltonian with 15 Pauli terms [33].
Ansatz Circuit: The quantum circuit is initialized to the Hartree-Fock state |1100⟩, followed by a
DoubleExcitationoperation (Givens rotation) that couples the |1100⟩ and |0011⟩ states with a single parameter θ [33].Measurement Protocol: The expectation value of each Pauli term is measured individually, requiring separate basis rotations for non-Z measurements. The results are combined with appropriate coefficients to compute the total energy [33].
Classical Optimization: A classical optimizer (e.g., SGD, SLSQP, or ADAM) adjusts the parameter θ to minimize the energy expectation value, typically requiring 10-100 iterations for convergence [33] [34].
Quantum Krylov Subspace Diagonalization
The experimental protocol for quantum Krylov methods involves [36]:
Krylov Basis Generation: Generate basis states by applying powers of the time-evolution operator U = e^(-iHτ) with carefully chosen time steps τ to an initial state |ψ₀⟩.
Overlap and Hamiltonian Matrix Construction: Measure all matrix elements Hᵢⱼ = ⟨ψᵢ|H|ψⱼ⟩ and Sᵢⱼ = ⟨ψᵢ|ψⱼ⟩ through quantum measurements. Recent MSD approaches reduce this measurement cost by expressing the Hamiltonian as a linear combination of time-evolution unitaries [35].
Generalized Eigenvalue Solution: Solve the generalized eigenvalue problem Hc = ESc classically to obtain energy estimates and state approximations within the Krylov subspace.
Error Mitigation: Apply regularization techniques to address ill-conditioned overlap matrices and statistical error analysis to account for measurement noise [36].
Quantum Phase Estimation Protocol
The standard QPE experimental procedure comprises [37]:
Register Initialization: Prepare the estimation register with n qubits in |0⟩ state and the state register with the initial state |ψ⟩, ideally with high overlap with the target eigenstate.
Superposition Creation: Apply Hadamard gates to all estimation qubits to create a uniform superposition.
Controlled-Unitary Operations: Apply controlled-U^(2^k) operations for k = 0 to n-1, where the unitary is typically U = e^(-iHt) for a suitably chosen t.
Inverse Fourier Transform: Apply the inverse Quantum Fourier Transform to the estimation register.
Measurement and Interpretation: Measure the estimation register in the computational basis and convert the binary result to a phase estimate θ = 0.θ₁θ₂...θₙ, yielding energy E = 2πθ/t.
For the Filtered QPE variant, an additional spectral filtering step is applied to the initial state to enhance overlap with the target eigenstate before the standard QPE procedure [39].
Visualization of Algorithmic Workflows
VQE Algorithmic Flow
Quantum Krylov Subspace Method
Quantum Phase Estimation Circuit
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Computational Tools for Quantum Ground State Calculations
| Tool/Component | Function | Example Implementations |
|---|---|---|
| Molecular Hamiltonians | Encodes electronic structure problem | PySCF [34], PennyLane[qchem [33]] |
| Qubit Mappers | Fermionic-to-qubit transformation | Jordan-Wigner [33], Bravyi-Kitaev [32], ParityMapper [34] |
| Ansatz Circuits | Parameterized trial wavefunctions | UCCSD [34], Hardware-Efficient [32], DoubleExcitation [33] |
| Classical Optimizers | Hybrid algorithm parameter optimization | SLSQP [34], SPSA [34], ADAM [34], Gradient Descent [33] |
| Quantum Simulators | Algorithm testing and validation | PennyLane [33] [37], Qiskit Aer [34], Cirq |
| Measurement Tools | Expectation value estimation | Estimator [34], Shot-based sampling [32] |
The three algorithmic paradigms—VQE, Quantum Krylov, and QPE—offer complementary approaches to the molecular ground state problem with distinct trade-offs. VQE provides immediate utility on current NISQ devices but faces challenges with optimization and convergence. Quantum Krylov methods offer improved convergence guarantees with moderate quantum resources, making them promising for early fault-tolerant devices. QPE remains the asymptotically optimal approach for fault-tolerant quantum computers, with recent FQPE variants addressing critical overlap limitations. For pharmaceutical researchers, the optimal algorithm choice depends on available quantum hardware, molecular system size, and required precision, with hybrid approaches potentially offering the most practical near-term path toward quantum utility in drug development.
The accurate computation of molecular ground-state energies is a fundamental challenge in quantum chemistry, with profound implications for drug design and materials science. Classical computational methods often struggle with the exponential scaling required to simulate large, strongly correlated quantum systems. Within the framework of quantum computing for molecular Hamiltonian ground state search, hybrid quantum-classical algorithms emerge as a pivotal strategy for the current noisy intermediate-scale quantum (NISQ) era. Among these, the integration of Density Matrix Embedding Theory (DMET) and Sample-Based Quantum Diagonalization (SQD) represents a significant advance, enabling accurate simulations of large molecules by strategically distributing the computational workload between quantum and classical processors [40] [41]. This guide provides an in-depth technical examination of the DMET-SQD methodology, its experimental implementation, and its application to problems of biological relevance.
Theoretical Foundations
Density Matrix Embedding Theory (DMET)
DMET is a quantum embedding technique that allows a large, intractable molecular system to be solved by dividing it into smaller, manageable fragments coupled to a mean-field environment [42] [41].
- Core Principle: The formally exact Schmidt decomposition allows the ground state of a full system to be expressed through an impurity model containing only 2N degrees of freedom for a fragment of size N, significantly reducing the problem's complexity [42].
- Self-Consistent Loop: An effective local potential (U) couples the fragment to its environment. This potential is optimized self-consistently by matching the density matrix of the impurity model and the effective lattice model projected onto the fragment [42].
- Role in Hybrid Computing: Within a quantum-centric supercomputing (QCSC) architecture, DMET provides the overarching framework for fragmenting the molecule, allowing the quantum processor to focus computational resources on the chemically relevant subsystem where electron correlation is strongest [40] [41].
Sample-Based Quantum Diagonalization (SQD)
SQD is a quantum diagonalization algorithm designed to approximate ground-state energies on near-term quantum devices without requiring deep parameter optimization [43] [44].
- Core Principle: A quantum device samples electronic configurations (bitstrings) from a prepared ansatz state. Classical high-performance computing (HPC) resources then post-process these samples to reconstruct a set of dominant configurations [44].
- Classical Diagonalization: The Hamiltonian is diagonalized within the subspace spanned by the recovered configurations, effectively solving the Schrödinger equation for a drastically reduced Hilbert space [44].
- Noise Resilience: As a sample-based method, SQD is inherently tolerant to the noise present on current quantum hardware, and its accuracy can be systematically improved by increasing the number of sampled configurations [43] [44].
Methodological Integration: The DMET-SQD Workflow
The integration of DMET and SQD creates a powerful synergy for simulating large molecules. The workflow below illustrates the self-consistent interaction between the classical DMET framework and the quantum SQD subroutine.
DMET-SQD Self-Consistent Workflow: The process initiates with a mean-field calculation for the full molecule. A fragment is selected, and its entangled quantum bath is constructed via Schmidt decomposition. The embedded Hamiltonian for this fragment-plus-bath system is formed and passed to the SQD subroutine on the quantum computer. The resulting fragment density matrix is fed back to the classical computer. The process iterates until self-consistency is achieved between the fragment density and the global mean-field potential [40] [41].
The SQD Subroutine
The quantum SQD subroutine, which solves the embedded Hamiltonian, follows a detailed sequence of steps as shown in the following workflow.
SQD Subroutine Detail: The embedded Hamiltonian is used to prepare an ansatz state (e.g., the Local Unitary Coupled Cluster - LUCJ ansatz) on the quantum processing unit (QPU). The QPU then samples electronic configurations from this state. These noisy samples are processed classically by the S-CORE algorithm to mitigate errors and recover the most important configurations [44]. The Hamiltonian and overlap matrices are built within this subspace and diagonalized on a classical HPC system to obtain the ground-state energy and density matrix for the fragment [43] [44].
Experimental Protocols & Validation
The DMET-SQD methodology has been experimentally validated on quantum hardware for specific molecular systems, demonstrating its potential for biomedical applications [40].
Key Experimental Demonstrations
Table 1: Summary of DMET-SQD Experimental Validations
| Molecular System | Qubits Used (Full / Fragment) | Quantum Hardware | Key Result | Classical Benchmark |
|---|---|---|---|---|
| Hydrogen Ring (18 atoms) | 41 / 27 | ibm_cleveland (Eagle processor) |
Agreement with HCI benchmarks [40] [41] | Heat-Bath CI (HCI) |
| Cyclohexane Conformers | 89 / 32 | ibm_cleveland (Eagle processor) |
Energy differences within 1 kcal/mol of CCSD(T) [40] [41] | CCSD(T) |
Detailed Protocol: Cyclohexane Conformer Calculation
The following workflow and protocol outline the steps for calculating relative conformer energies, a critical test for drug development where molecular conformation dictates biological activity.
- System Preparation: The molecular geometry of each cyclohexane conformer (chair, boat, twist-boat, half-chair) is first optimized using classical computational methods [40].
- Active Space Selection: For each conformer, a chemically relevant fragment (e.g., a set of carbon atoms and their associated electrons) is selected. The DMET procedure maps the full 89-qubit problem to a 32-qubit embedded Hamiltonian [41].
- Quantum Execution: The SQD subroutine is executed on the
ibm_clevelandquantum processor for the 32-qubit fragment. This involves: - Classical Post-Processing: The sampled configurations are used to construct and diagonalize the generalized eigenvalue problem on a classical HPC cluster.
- Self-Consistency: The fragment density matrix is fed back into the DMET loop until convergence is achieved for the total energy.
- Validation: The relative energies between conformers computed via DMET-SQD are compared to established classical methods like CCSD(T) and HCI, with successful results falling within the threshold of chemical accuracy (1 kcal/mol) [40].
The Scientist's Toolkit: Research Reagent Solutions
Implementing the DMET-SQD workflow requires a suite of software and hardware components.
Table 2: Essential Research Tools for DMET-SQD Simulations
| Tool Name | Type | Primary Function | Application in DMET-SQD |
|---|---|---|---|
| Tangelo [40] | Software Library | Quantum Chemistry Toolkit | Provides the DMET framework and interfaces with quantum software. |
| Qiskit / SQD Addon [44] | Software Library | Quantum Algorithm Development | Implements the SQD algorithm, including configuration sampling and recovery. |
| PySCF [44] | Software Library | Classical Electronic Structure | Performs initial mean-field calculations and integral transformation. |
| AVAS [44] | Software Tool | Automated Active Space Selection | Identifies optimal molecular orbitals for the fragment, streamlining setup. |
IBM Quantum Hardware (e.g., ibm_cleveland) |
Quantum Hardware | Quantum Processing Unit (QPU) | Executes the quantum circuits for the SQD subroutine [40] [41]. |
| Classical HPC Cluster | Classical Hardware | High-Performance Computing | Handles classical pre- and post-processing, including diagonalization of large subspaces [44]. |
Applications and Future Outlook
The integration of DMET and SQD marks a tangible step toward practical quantum-centric simulations for drug discovery. By accurately computing the relative energies of molecular conformers and simulating non-covalent interactions, this approach can impact the early stages of drug design, where understanding protein-ligand binding and molecular stability is critical [40] [44].
Future work will focus on refining the method to overcome current limitations, such as the use of minimal basis sets and the accuracy of sampling in strongly correlated systems [40]. As quantum hardware improves with lower error rates and higher qubit counts, DMET-SQD is poised to simulate increasingly complex biological systems, such as peptides and proteins, potentially unlocking new paths to medical advances [41].
In the pursuit of simulating quantum molecular systems to accelerate drug discovery and materials design, finding the ground state energy of a molecular Hamiltonian is a fundamental challenge. Classical computational methods, such as those based on the Coupled Cluster theory, struggle with the exponential scaling of the Hilbert space for systems exhibiting strong electron correlations. Quantum computing offers a promising pathway to overcome this hurdle. This technical guide details two core procedural pillars essential for quantum algorithms targeting molecular ground states: the preparation of problem-specific ansätze and the decomposition of the molecular Hamiltonian into measurable Pauli operators. Framed within the context of variational quantum algorithms, these techniques enable the use of both near-term noisy intermediate-scale quantum (NISQ) devices and future fault-tolerant quantum computers for quantum chemistry problems [4] [45].
Matrix (Ansatz) Preparation Techniques
The preparation of a parameterized quantum state, or ansatz, is a critical step in variational algorithms like the Variational Quantum Eigensolver (VQE). The choice of ansatz significantly influences the convergence, accuracy, and resource requirements of the ground state search.
Adaptive Ansatz Construction: ADAPT-VQE and Beyond
Adaptive algorithms dynamically build a problem-specific ansatz, offering a compact and expressive alternative to fixed-ansatz approaches.
- ADAPT-VQE: The standard ADAPT-VQE algorithm iteratively grows an ansatz by selecting operators from a predefined pool (e.g., based on Unitary Coupled-Cluster single and double excitations, UCCSD). At each iteration, it calculates the energy gradient with respect to each operator in the pool and selects the one with the largest gradient magnitude for inclusion in the circuit. This process continues until the energy converges to a desired accuracy, such as chemical accuracy [4].
- K-ADAPT-VQE: An enhancement to ADAPT-VQE, this method improves efficiency by adding multiple (
K) operators to the ansatz in each iteration, a process referred to as "chunking." This strategy substantially reduces the total number of circuit evaluations and optimization iterations required to achieve chemical accuracy, as demonstrated in simulations of small molecules like BeH₂, LiH, and N₂ [4]. - ADAPT-AQC: This algorithm uses an adaptive-ansatz approach for the related task of preparing a specific target state, such as a Matrix Product State (MPS). It dynamically constructs a circuit by iteratively placing two-qubit unitaries based on a selection criterion, followed by parameter optimization. This method has been shown to prepare complex states like molecular ground states and the ground state of a 50-site Heisenberg model with high fidelity [46].
Matrix Product State (MPS) Based Preparation
Matrix Product States provide a structured, efficient representation for quantum states with limited entanglement, making them suitable for both classical simulation and quantum state preparation.
- Efficient Representation: An MPS represents an
n-qubit state usingnconnected tensors. The maximum dimension of the connections, known as the bond dimension (χ), controls the amount of entanglement the MPS can capture. States with low entanglement can be accurately represented with a smallχ, leading to an efficient classical description [47]. - State Preparation via Compression: Any quantum state can be decomposed into an MPS using a sequence of Singular Value Decompositions (SVDs). This process involves iteratively reshaping the state vector into a matrix, performing an SVD, and retaining only the largest
χsingular values and corresponding vectors. This truncation compresses the state, and the resulting components form the MPS tensors [47]. The resulting MPS can then be compiled into a quantum circuit. - Application to Smooth Distributions: Probability distributions that are smooth and differentiable can be efficiently approximated by an MPS with low bond dimension, as their entanglement grows slowly with system size. This property is leveraged to prepare quantum states that encode normal distributions, which are useful in quantum algorithms like Monte-Carlo methods [48].
Compiled and Variational Preparation
For near-term hardware, shallow circuits are paramount. AQC-Tensor is an algorithm that variationally optimizes a parameterized brickwork circuit to approximate a target MPS. Its performance is greatly enhanced by initializing the circuit to prepare a compressed version (χ=1) of the target state, which provides better initial fidelity and gradients for the classical optimizer compared to random initialization [46].
The table below summarizes the core matrix preparation techniques.
Table 1: Core Matrix (Ansatz) Preparation Techniques
| Technique | Core Principle | Key Advantage | Primary Application |
|---|---|---|---|
| ADAPT-VQE | Iterative, gradient-based selection of operators from a pool [4]. | Compact, problem-specific circuit. | Molecular ground state energy calculation. |
| K-ADAPT-VQE | Adds K operators per iteration ("chunking") [4]. |
Reduced quantum resource overhead. | Molecular ground state energy calculation. |
| MPS SVD Compression | Truncated SVD to decompose a state into a tensor network [47]. | Efficient classical representation of low-entanglement states. | General state preparation; loading smooth distributions [48]. |
| ADAPT-AQC | Adaptive, heuristic placement of two-qubit gates [46]. | Shallow circuits for specific state classes (e.g., normal MPS). | Preparing MPS representations of molecular and Heisenberg ground states. |
| AQC-Tensor | Variational optimization of a fixed brickwork ansatz [46]. | Shallow, hardware-friendly circuits. | Preparing MPS representations of molecular and Heisenberg ground states. |
Experimental Protocol: K-ADAPT-VQE for a Molecular System
The following workflow details the steps for conducting a ground state search using the K-ADAPT-VQE algorithm.
Figure 1: K-ADAPT-VQE ground state search workflow, showing the hybrid quantum-classical loop [4].
- Problem Formulation: Define the molecular system (e.g., LiH, BeH₂, N₂) and its nuclear coordinates at a specific bond length [4].
- Classical Pre-processing:
- Use a quantum chemistry package like PySCF to perform a Hartree-Fock calculation on the defined molecule. This provides the molecular orbital coefficients and a reference state (
|Ψ_HF〉) [4]. - Compute the one- and two-electron integrals to construct the second-quantized electronic Hamiltonian.
- Employ a library like OpenFermion to map the fermionic Hamiltonian to a qubit Hamiltonian using a transformation such as Jordan-Wigner [4].
- Use a quantum chemistry package like PySCF to perform a Hartree-Fock calculation on the defined molecule. This provides the molecular orbital coefficients and a reference state (
- Algorithm Initialization:
- Prepare the Hartree-Fock state on the quantum processor.
- Define the operator pool, typically consisting of all symmetry-allowed fermionic excitation operators (e.g., from UCCSD theory) mapped to qubit operators [4].
- Iterative Ansatz Construction:
- Gradient Calculation: For each operator in the pool, estimate the energy gradient (e.g.,
∂E/∂θ_i) using the quantum computer. - Operator Selection: Identify the
Koperators with the largest gradient magnitudes [4]. - Ansatz Growth: Append the corresponding parameterized gates for these
Koperators to the quantum circuit. - Parameter Optimization: Use a classical optimizer (e.g., COBYLA or CMA-ES) to minimize the energy expectation value with respect to all parameters in the current ansatz. This step involves repeated calls to the quantum device for energy estimation [4].
- Gradient Calculation: For each operator in the pool, estimate the energy gradient (e.g.,
- Termination: The algorithm iterates until the energy difference between successive steps falls below a predefined threshold, such as the chemical accuracy target (∼1.6 mHa).
Pauli Decomposition and Measurement
The electronic structure Hamiltonian, generated in second quantization, must be translated into operations native to a quantum computer. This involves decomposing it into a sum of Pauli operators.
Hamiltonian Formulation and Mapping
The molecular Hamiltonian in second quantization is: [ \hat{H} = \sum{pq} h{pq} \hat{a}p^\dagger \hat{a}q + \frac{1}{2} \sum{pqrs} h{pqrs} \hat{a}p^\dagger \hat{a}q^\dagger \hat{a}r \hat{a}s ] where ( h{pq} ) and ( h{pqrs} ) are one- and two-electron integrals, and ( \hat{a}^\dagger ) and ( \hat{a} ) are fermionic creation and annihilation operators [4]. To run this on a quantum computer, it must be mapped to qubits. The Jordan-Wigner transformation is a common choice, which maps fermionic operators to Pauli strings (tensor products of ( I, X, Y, Z )) while preserving the anti-commutation relations [4]. The result is a qubit Hamiltonian of the form: [ H = \sum{i} ci Pi ] where ( ci ) are real coefficients and ( P_i ) are Pauli strings.
Pauli Measurement Techniques
Measuring the energy expectation value ( \langle H \rangle = \sumi ci \langle Pi \rangle ) requires estimating the expectation value of each Pauli term ( \langle Pi \rangle ). This is not trivial, as many Pauli operators do not correspond to a simple computational basis measurement.
- Single-Qubit Pauli Measurement: Measuring a single-qubit Pauli operator is equivalent to performing a rotation before a computational basis (
Z) measurement [49].- To measure
X, apply the Hadamard gateHbefore measurement. - To measure
Y, applyS†followed byHbefore measurement [49].
- To measure
- Multi-Qubit Pauli Measurement: Measuring a multi-qubit Pauli operator like
X⊗ZorZ⊗Zinvolves measuring the parity between qubits. This is typically accomplished by using a basis-changing rotation followed by entangling gates, such as CNOT, to propagate parity information to a single ancilla or target qubit, which is then measured in theZbasis [49]. For example, measuringZ⊗Zcan be done by applying a CNOT from the first to the second qubit, then measuring the second qubit in theZbasis. The outcome corresponds to the parity of the two qubits [49].
Table 2: Single-Qubit Pauli Measurement Protocol [49]
| Pauli Operator | Unitary Transformation | Q#-Style Pseudocode |
|---|---|---|
| ( Z ) | ( I ) | M(qubit) |
| ( X ) | ( H ) | H(qubit); M(qubit); |
| ( Y ) | ( H S^\dagger ) | Adjoint S(qubit); H(qubit); M(qubit); |
Measurement Optimization
Since each Pauli term ( P_i ) might require a different measurement basis, a naive approach of measuring each term separately would be prohibitively expensive. Measurement grouping is a critical optimization technique where Pauli terms that are diagonal in the same tensor product basis (i.e., that commute and can be measured with the same pre-rotation circuit) are grouped together and measured simultaneously. This significantly reduces the number of distinct quantum measurements required.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Software and Algorithmic Tools for Hamiltonian Simulation Research
| Item | Function | Application in Workflow |
|---|---|---|
| PySCF | Open-source quantum chemistry package; performs Hartree-Fock calculations and computes molecular integrals [4]. | Classical pre-processing for Hamiltonian generation. |
| OpenFermion | Library for compiling and analyzing quantum algorithms for chemistry; transforms fermionic operators to qubit operators [4]. | Qubit mapping (e.g., via Jordan-Wigner). |
| UCCSD Operator Pool | A predefined set of unitary coupled-cluster single and double excitation operators [4]. | Forms the operator pool for adaptive ansatz construction in (K-)ADAPT-VQE. |
| Matrix Product State (MPS) | A tensor network formalism for efficiently representing quantum states with limited entanglement [47]. | Classical compression and representation of target states; basis for variational state preparation algorithms [46]. |
| Singular Value Decomposition (SVD) | A matrix factorization method used for dimensionality reduction and compression [47]. | Core linear algebra routine for constructing and compressing MPS. |
| Jordan-Wigner Transformation | A specific technique for mapping fermionic creation/annihilation operators to Pauli spin operators [4]. | Encoding the fermionic molecular Hamiltonian into a qubit-readable form. |
The successful implementation of quantum algorithms for molecular ground state search hinges on the sophisticated interplay of advanced matrix preparation techniques and efficient Pauli decomposition protocols. Adaptive ansätze like K-ADAPT-VQE offer a path to compact, problem-tailored quantum circuits, while MPS-based methods provide a powerful framework for preparing states with specific properties, such as those with limited entanglement or encoding smooth probability distributions. The decomposition of the complex molecular Hamiltonian into measurable Pauli terms, coupled with strategic measurement optimizations, forms the bridge between theoretical chemistry and executable quantum circuits. As quantum hardware continues to mature, the refinement of these core techniques—particularly in reducing circuit depth and minimizing measurement overhead—will be crucial for achieving a quantum advantage in simulating molecular systems for drug development and materials science.
Efficiently preparing the ground state of a molecular Hamiltonian is a critical task in quantum computation for chemistry, with direct applications in drug development and materials science [50] [4]. The challenge lies in transforming a simple, easy-to-prepare initial quantum state into a complex ground state that accurately represents the electronic structure of a molecule. This process is a significant bottleneck; the time required for state preparation can often exceed the runtime of the quantum algorithm itself, making efficiency paramount [51]. This technical guide provides an in-depth examination of the dominant state preparation strategies, focusing on the core methodologies of adiabatic evolution and variational approaches, with a specific focus on their application to molecular systems.
Core Methodological Frameworks
Adiabatic State Preparation (ASP)
Adiabatic State Preparation is a heuristic algorithm grounded in the adiabatic theorem of quantum mechanics [50]. The core principle involves a continuous transformation from a simple initial Hamiltonian, whose ground state is known and easily preparable, to a final target Hamiltonian, whose ground state one wishes to prepare.
The system is evolved under a time-dependent Hamiltonian, typically a linear interpolation: [ \hat{H}(s) = (1-s)\hat{H}{\mathrm{i}} + s\hat{H}{\mathrm{f}}, \quad 0 \leq s \leq 1 ] where ( \hat{H}{\mathrm{i}} ) is the initial Hamiltonian, ( \hat{H}{\mathrm{f}} ) is the final target Hamiltonian, and ( s = t/T{\text{ASP}} ) is a dimensionless time parameter for a total evolution time ( T{\text{ASP}} ) [50]. According to the adiabatic theorem, if the system begins in the ground state of ( \hat{H}{\mathrm{i}} ) and the evolution is sufficiently slow relative to the inverse square of the minimum energy gap ( \gamma(s) = \lambda1(s) - \lambda_0(s) ) between the ground and first excited states, the system will remain in the instantaneous ground state throughout the evolution.
A key quantitative measure for the stability of the adiabatic path is the adiabatic estimate: [ T{\text{est}} = \max{0 \leq s \leq 1} \max{j>0} \frac{\left| \langle \phij(s) | \frac{d\hat{H}}{ds}(s) | \phi0(s) \rangle \right|}{(\lambdaj(s) - \lambda_0(s))^2} ] This quantity, which can be numerically evaluated, helps benchmark different adiabatic paths and is independent of the scheduling function [50].
A significant advancement is the development of methods to construct an adiabatic path from a generic initial wavefunction, not just simple product states [50]. Given a quantum circuit that prepares an initial trial wavefunction ( |\psiT\rangle ), one can perform measurements to approximate a parent Hamiltonian ( \hat{H}{\mathrm{P}} ) for which ( |\psi_T\rangle ) is a ground state. This allows any efficiently preparable state, including outputs from other quantum algorithms, to serve as the starting point for an adiabatic evolution to the true target ground state [50].
Variational Quantum Algorithms
Variational algorithms, such as the Variational Quantum Eigensolver (VQE), take a hybrid quantum-classical approach to ground-state preparation [4]. A parametrized quantum circuit (ansatz) prepares a trial state ( |\Psi(\vec{\theta})\rangle ) on the quantum processor. The energy expectation value ( E(\vec{\theta}) = \langle \Psi(\vec{\theta}) | \hat{H} | \Psi(\vec{\theta}) \rangle ) is measured, and a classical optimizer adjusts the parameters ( \vec{\theta} ) to minimize this energy.
The Adaptive Derivative-Assembled Pseudo-Trotter (ADAPT-VQE) algorithm improves upon standard VQE by dynamically constructing a compact, problem-specific ansatz [4]. It starts from an initial state (e.g., the Hartree-Fock state) and iteratively adds unitary operators from a predefined pool (e.g., UCCSD excitations), selecting the operator that yields the largest gradient toward lowering the energy.
The K-ADAPT-VQE variant enhances efficiency by adding K operators with the largest gradients to the ansatz in each iteration, a process known as "chunking" [4]. This strategy significantly reduces the total number of quantum function evaluations and VQE iterations required to achieve chemical accuracy, mitigating the computational overhead associated with the original adaptive algorithm.
The Piecewise Quantum Singular Value Transformation
The Quantum Singular Value Transformation (QSVT) is a powerful framework for constructing polynomial transformations of matrices encoded in quantum circuits. The recently introduced piecewise QSVT extends this capability to states whose amplitudes can be well-approximated by piecewise polynomials [51].
This technique facilitates the preparation of complex states that contain sharp discontinuities or singularities, which are challenging for other methods. It works by applying different polynomial transformations to different regions of the input data, supported by a new piecewise linear diagonal block encoding [51]. This approach has been shown to efficiently prepare states like ( x^\alpha |x\rangle ) and ( \log x |x\rangle ), and has a direct application in improving Quantum Phase Estimation (QPE) by preparing optimized B-spline window states with a reported 50-fold reduction in the number of Toffoli gates compared to the state-of-the-art Kaiser window state [51].
Comparative Analysis of Strategies
The following table summarizes the key characteristics, requirements, and recent advancements for each state preparation strategy.
Table 1: Comparative Analysis of Quantum State Preparation Strategies
| Strategy | Core Principle | Key Requirements | Recent Advancements | Reported Performance/Resource Estimates |
|---|---|---|---|---|
| Adiabatic (ASP) | Slow interpolation between initial and target Hamiltonians [50]. | Knowledge of initial ground state; gapped path; long coherent time. | Parent Hamiltonian construction from generic initial states [50]. | Performance depends on ( T_{\text{est}} ); applied to H₂O and CH₂ [50]. |
| Variational (K-ADAPT-VQE) | Hybrid optimization of parameterized quantum circuit [4]. | Efficient ansatz; classical optimizer; robust measurement. | Operator "chunking" (K-ADAPT); operator pool pruning [4]. | Reduces quantum function calls & iterations to achieve chemical accuracy for BeH₂, LiH, N₂ [4]. |
| Piecewise QSVT | Application of piecewise polynomial transformations via block encoding [51]. | Block encoding of target function; polynomial approximation. | Framework for states with piecewise polynomial amplitudes [51]. | B-spline window preparation uses 50x fewer Toffolis vs. Kaiser window [51]. |
Experimental Protocols
Protocol: Adiabatic State Preparation with a Parent Hamiltonian
This protocol details the steps for performing ASP starting from a generic initial state, as described in the recent work by Fullera et al. [50].
Input Preparation:
- Target Hamiltonian (( \hat{H}_{\mathrm{f}} )): The molecular Hamiltonian of interest, derived in a qubit basis via a tool like OpenFermion [4].
- Initial Trial State (( |\psi_T\rangle )): A quantum circuit that prepares a generic initial wavefunction, which could be the output of another algorithm (e.g., a shallow VQE circuit or a Hartree-Fock state).
Parent Hamiltonian Construction:
- Perform a series of measurements on ( |\psi_T\rangle ) to gather information about its expectation values.
- Use this data to heuristically construct (or approximate) a parent Hamiltonian ( \hat{H}{\mathrm{P}} ) for which ( |\psiT\rangle ) is an eigenstate. The method sometimes identifies a continuous subspace of valid parent Hamiltonians, allowing for optimization based on sparsity or similarity to ( \hat{H}_{\mathrm{f}} ) [50].
- The quality of this parent Hamiltonian approximation can be checked prior to running the full ASP algorithm.
Adiabatic Path Definition:
- Define the adiabatic path, for example, via linear interpolation: [ \hat{H}(s) = (1-s)\hat{H}{\mathrm{P}} + s\hat{H}{\mathrm{f}} ]
Path Benchmarking (Pre-Run):
- Discretize the parameter
sand numerically estimate the adiabatic estimate ( T_{\text{est}} ) from Eq. (6) to benchmark the difficulty of the path [50].
- Discretize the parameter
State Evolution:
- Prepare the initial state ( |\psi_T\rangle ) on the quantum computer.
- Evolve the system under the time-dependent Hamiltonian ( \hat{H}(s) ) for a total time ( T{\text{ASP}} ), chosen to be significantly larger than ( T{\text{est}} ) to satisfy the adiabatic condition.
Verification:
- Measure the energy of the final state ( |\Psi(1)\rangle ) with respect to ( \hat{H}_{\mathrm{f}} ) to verify proximity to the ground-state energy.
Protocol: K-ADAPT-VQE for Molecular Ground States
This protocol outlines the K-ADAPT-VQE method for finding molecular ground states [4].
Input Preparation:
- Molecular Geometry: Define the atomic species and their coordinates.
- Basis Set: Choose a molecular orbital basis (e.g., STO-3G).
- Active Space: Select an active space for the calculation if necessary.
Hamiltonian and Operator Pool Generation:
- Use a quantum chemistry package (e.g., PySCF) to perform a Hartree-Fock calculation and compute one- and two-electron integrals [4].
- Use a library like OpenFermion to construct the fermionic Hamiltonian and map it to a qubit Hamiltonian using a transformation such as Jordan-Wigner [4].
- Construct the operator pool, typically comprised of all symmetry-allowed single and double fermionic excitation operators (( \hat{T}1, \hat{T}2 )) from the UCCSD ansatz, then map them to qubit operators [4].
Algorithm Execution:
- Initialization: Prepare the Hartree-Fock state ( |\Psi_{\text{HF}}\rangle ) on the quantum processor. Initialize the ansatz circuit as empty.
- Iterative Loop:
a. Gradient Calculation: For each operator ( \hat{A}i ) in the operator pool, measure the energy gradient component ( gi = \langle \Psi | [\hat{H}, \hat{A}i] | \Psi \rangle ).
b. Operator Selection: Select the
Koperators with the largest absolute gradient magnitudes. c. Ansatz Update: Append theseKoperators (as parameterized unitaries, e.g., ( e^{-i\thetai \hat{A}_i} )) to the ansatz circuit. d. Parameter Optimization: Use a classical optimizer (e.g., COBYLA, CMA-ES) to minimize the energy with respect to all parameters in the expanded ansatz. - Convergence Check: The loop terminates when the norm of the gradient vector falls below a predefined threshold, indicating that a local minimum has been reached.
Essential Research Toolkit
For researchers implementing these protocols, the following tools and concepts are indispensable.
Table 2: The Scientist's Toolkit: Essential Reagents and Resources
| Item | Function/Description | Example Use-Case |
|---|---|---|
| Molecular Hamiltonian | The target operator whose ground state is sought; defines the physical system [4]. | Core input for all state preparation algorithms. |
| Hartree-Fock State | A mean-field approximation of the molecular ground state; a common, simple initial state [4]. | Starting point for VQE/ADAPT-VQE and some ASP evolutions. |
| UCCSD Operator Pool | A set of unitary excitation operators used to build correlated ansätze [4]. | Forms the operator pool in the (K-)ADAPT-VQE algorithm. |
| Parent Hamiltonian | A Hermitian operator constructed to have a specific state as its eigenstate [50]. | Enables ASP to start from a high-quality, generic initial state. |
| Block Encoding | A technique for embedding a non-unitary matrix as a sub-block of a larger unitary matrix [51]. | Fundamental subroutine for implementing the piecewise QSVT protocol. |
| B-spline Window State | An optimized initial state for Quantum Phase Estimation with excellent spectral properties [51]. | Used in QPE to reduce Trotter error; efficiently preparable via piecewise QSVT. |
Workflow and System Diagrams
The following diagrams illustrate the logical flow of the key algorithms discussed.
K-ADAPT-VQE Algorithm Flow
Adiabatic State Preparation Workflow
The field of quantum state preparation is rapidly advancing, moving beyond generic methods to highly specialized and efficient protocols. Adiabatic State Preparation remains a powerful, conceptually clear method, now enhanced by techniques that allow it to start from sophisticated initial states via parent Hamiltonian construction [50]. Variational approaches, particularly adaptive algorithms like K-ADAPT-VQE, offer a resource-efficient pathway on near-term hardware by dynamically tailoring the ansatz and reducing iteration counts [4]. Meanwhile, advanced techniques like piecewise QSVT open the door to preparing previously challenging states with complex functional forms, offering dramatic resource savings for key subroutines like Quantum Phase Estimation [51].
For researchers in quantum chemistry and drug development, the choice of strategy depends on the specific molecular system, the available quantum hardware, and the desired balance between circuit depth and classical computation. The ongoing development of these methods is crucial for realizing the potential of quantum computing in simulating complex molecular systems.
The discovery of novel therapeutics for complex biological targets represents one of the most computationally intensive challenges in modern science. Two particularly difficult classes of targets—the KRAS oncogene and metalloenzymes such as metallo-β-lactamases (MBLs)—have long eluded conventional drug development approaches due to the quantum mechanical complexities underlying their function. These challenges frame a compelling application for quantum computing, specifically for molecular Hamiltonian ground state search algorithms. The molecular Hamiltonian (Ĥ) encapsulates the total energy of a quantum system, and finding its ground state energy (E₀), defined as E₀ = min|Ψ⟩⟨Ψ|Ĥ|Ψ⟩, is critical for predicting molecular stability, reactivity, and binding interactions [52] [53].
This technical guide examines how quantum computing approaches are being deployed to overcome the limitations of classical computational methods in drug discovery. We explore two specific case studies: the covalent inhibition of the KRAS G12C mutant and the inhibition of metallo-β-lactamases to combat antibiotic resistance. For each, we provide detailed methodologies, computational protocols, and resources that integrate quantum Hamiltonian simulation techniques into practical research pipelines.
Case Study I: Targeting the KRAS Oncogene
Biological and Clinical Significance
The KRAS protein is a crucial molecular switch in cellular signaling pathways that controls cell growth, differentiation, and survival. As a GTPase, it cycles between an active GTP-bound state and an inactive GDP-bound state. Oncogenic mutations, particularly at codon G12, impair its GTPase activity, leaving the protein constitutively active and driving uncontrolled cell proliferation [54] [55]. KRAS mutations are foundational to in vivo oncogenic transformation and are highly prevalent in several lethal cancers, including pancreatic ductal adenocarcinoma (PDAC) (~90%), colorectal cancer (~40%), and non-small cell lung cancer (NSCLC) (~21%) [54].
The specific mutation variants display distinct tissue distributions, which is critical for therapeutic targeting. G12D is the most prevalent variant in PDAC (approximately 37-45% of cases), while G12C is most common in NSCLC (13.6% of cases) [54] [56]. For decades, KRAS was considered "undruggable" due to its smooth surface and picomolar affinity for GTP, which made designing competitive inhibitors exceptionally challenging [54] [55].
Table 1: Prevalence of Major KRAS Mutations in Solid Tumors [54]
| Cancer Type | Overall KRAS Mutation Prevalence | Most Common Mutant Subtypes |
|---|---|---|
| Pancreatic Ductal Adenocarcinoma | 82.1% | G12D (37.0%), G12V (~22%), G12R |
| Colorectal Cancer | ~40% | G12D (12.5%), G12V (8.5%) |
| Non-Small Cell Lung Cancer | 21.2% | G12C (13.6%) |
Approved Therapeutics and Clinical Candidates
The breakthrough in targeting KRAS came with the discovery of allosteric pockets, particularly the switch-II pocket (S-IIP) in the KRAS G12C mutant. This allows for covalent binding with specific inhibitors that trap the protein in its inactive, GDP-bound state [54]. Two direct inhibitors, sotorasib (AMG510) and adagrasib (MRTX849), have received FDA approval for treating NSCLC harboring the G12C mutation [54] [53]. However, their efficacy is limited, with response rates of 30-40% and median progression-free survival of approximately 6 months, highlighting the challenge of resistance [54].
Research has since expanded to target other prevalent KRAS mutations. The current clinical pipeline includes two promising classes of inhibitors:
- RAS(ON)/Multi-KRAS Inhibitors: Daraxonrasib (RMC-6236) is designed to bind to and inhibit active, GTP-bound RAS, targeting several common KRAS mutations simultaneously. Early-phase trials have shown tumor shrinkage in patients with previously treated PDAC, leading to an ongoing global Phase 3 trial [56].
- Direct KRAS-G12D Inhibitors: MRTX1133 is a first-in-class drug designed to directly block the KRAS-G12D mutation, which is dominant in PDAC. It is currently in a Phase 1/2 trial (NCT05737706) for multiple solid tumors [56].
Table 2: Key KRAS-Targeted Therapeutics in Development [54] [56] [55]
| Therapeutic Agent | Target / Mechanism | Clinical Stage (as of 2025) | Key Cancer Types |
|---|---|---|---|
| Sotorasib (AMG510) | Covalent KRAS G12C inhibitor (inactive state) | FDA-approved (NSCLC) | NSCLC, Colorectal Cancer |
| Adagrasib (MRTX849) | Covalent KRAS G12C inhibitor (inactive state) | FDA-approved (NSCLC) | NSCLC, Colorectal Cancer |
| Daraxonrasib (RMC-6236) | RAS(ON) multi-KRAS inhibitor | Phase 3 | Pancreatic, NSCLC, Colorectal |
| MRTX1133 | Direct KRAS-G12D inhibitor | Phase 1/2 | Pancreatic, Colorectal |
Quantum Computing Application: Simulating Covalent Inhibition
The interaction between covalent inhibitors like sotorasib and the KRAS G12C protein involves complex quantum phenomena such as electron sharing and bond formation, which are poorly described by classical simulation methods like Molecular Mechanics (MM) [53]. A hybrid quantum-classical pipeline has been developed to enhance the accuracy of these simulations.
The workflow employs a QM/MM (Quantum Mechanics/Molecular Mechanics) scheme where the covalent binding site (the inhibitor and key cysteine residue of KRAS G12C) is treated quantum mechanically, while the rest of the protein and solvent environment is handled with classical molecular mechanics. The core quantum task is to solve the electronic structure problem for the QM region to calculate binding energies and interaction forces [53].
Detailed Protocol: QM/MM Simulation of KRAS G12C - Sotorasib Binding
System Preparation:
- Obtain the atomic coordinates of the KRAS G12C protein in complex with sotorasib from a protein data bank (PDB).
- Parameterize the system using a classical force field (e.g., AMBER, CHARMM). The sotorasib molecule and the cysteine residue at position 12 require special parameterization for the covalent bond.
- Solvate the protein-ligand complex in a water box and add ions to neutralize the system.
Region Partitioning:
- Define the QM region to include the sotorasib molecule and the side chain of cysteine 12. The MM region comprises the remainder of the protein, water, and ions.
- Set up the electronic structure calculation for the QM region. The Hamiltonian (Ĥ) is defined by the electrons and nuclei in the QM region, plus the electrostatic potential from the partial charges of the MM region.
Ground State Energy Calculation via VQE:
- Map the fermionic Hamiltonian of the QM region to a qubit Hamiltonian using a transformation such as Jordan-Wigner or parity.
- Choose a parameterized quantum circuit (ansatz), for instance, a hardware-efficient
R_yansatz with entangling gates. - On a quantum processor, measure the energy expectation value
⟨ψ(θ)|Ĥ|ψ(θ)⟩for a given set of parameters θ. - Use a classical optimizer (e.g., COBYLA, SPSA) in a feedback loop to minimize the energy expectation value. The output is the variational ground state energy E₀ and the corresponding wavefunction |ψ₀⟩.
Force Calculation and Geometry Optimization:
- Calculate the forces on the QM atoms' nuclei by evaluating the expectation value of the derivative of the Hamiltonian with respect to nuclear coordinates:
F = -⟨ψ₀|∂Ĥ/∂R|ψ₀⟩. - Use these quantum-computed forces to perform geometry optimization or molecular dynamics simulations of the binding pocket, leading to a more accurate prediction of the binding mode and affinity [53].
- Calculate the forces on the QM atoms' nuclei by evaluating the expectation value of the derivative of the Hamiltonian with respect to nuclear coordinates:
Diagram 1: QM/MM workflow for KRAS-inhibitor simulation
Case Study II: Targeting Metallo-β-Lactamases (MBLs)
Biological and Clinical Significance
Metallo-β-lactamases (MBLs) are bacterial zinc-dependent enzymes that confer resistance to a broad range of β-lactam antibiotics, including carbapenems, which are often last-line treatments for multidrug-resistant infections [57]. The rise of carbapenem-resistant Enterobacterales poses a severe threat to global public health. Unlike serine-β-lactamases (SBLs), for which clinical inhibitors exist, there are currently no approved MBL inhibitors (MBLIs), making the development of effective MBLIs a critical priority in the fight against antimicrobial resistance (AMR) [57].
The primary challenge in drug development for MBLs lies in their diverse active site architecture and the presence of one or two zinc ions, which are essential for the hydrolysis of the β-lactam ring in antibiotics. The metal ions play a crucial role in the catalytic mechanism, and designing inhibitors that can effectively chelate these ions without causing off-target toxicity has proven difficult [57].
Clinical Candidate and Discovery Challenges
The developmental pipeline for MBLIs is sparse, underscoring the difficulty of the problem. As of 2025, taniborbactam (VNRX-5133), a cyclic boronate-based broad-spectrum β-lactamase inhibitor, is in the pre-registration phase. It is administered in combination with cefepime and inhibits Ambler class A, C, and D serine enzymes, as well as class B MBLs like NDM and VIM [57]. Another promising candidate, xeruborbactam (QPX7728), is in Phase 1 clinical trials and targets an even broader spectrum of β-lactamases, including IMP MBLs [57].
A significant hurdle in MBLI discovery is the lack of standardized pre-clinical evaluation protocols. The field suffers from disparities in assay conditions, enzyme sources, and reporting methods, which stymie the comparison and reproducibility of potential inhibitors identified in different laboratories [57].
Quantum Computing Application: Simulating Zinc-Ion Interactions
Accurately modeling the interaction between potential inhibitors and the zinc ions in the MBL active site is a quantum problem. Classical force fields often poorly describe transition metal ions like zinc, particularly their coordination geometry, charge transfer, and dynamic bond formation. Quantum computing simulations can, in principle, provide a more accurate description of the electronic structure of these metal centers.
Detailed Protocol: Active Site Simulation for MBLI Design
Active Site Model Construction:
- Extract the MBL active site from a crystal structure, including the zinc ion(s), coordinating residues (e.g., Histidine, Aspartate, Cysteine), and crystallographic water molecules.
- Saturate any dangling bonds with hydrogen atoms to create a chemically valid molecular model for quantum chemical treatment.
Hamiltonian Formulation and Active Space Selection:
- Define the molecular Hamiltonian (Ĥ) for the active site model, which includes the kinetic energy of electrons and nuclei, as well as all coulombic interactions between them.
- The key challenge is selecting a chemically relevant "active space" for the calculation. This involves identifying a set of molecular orbitals and the electrons within them that are most relevant to the bonding and reactivity—typically the d-orbitals of the zinc ion(s) and the surrounding ligand orbitals. This is defined by the number of electrons and orbitals, e.g., a (4e, 4o) active space.
Ground State Energy Calculation via Phase Estimation or VQE:
- For near-term quantum devices, use the VQE algorithm as described in the KRAS protocol to find the ground state energy of the active site model.
- For fault-tolerant future quantum computers, the Quantum Phase Estimation (QPE) algorithm can be used to obtain the exact ground state energy without the approximations inherent in VQE.
- The calculation must be performed for the MBL active site alone, the inhibitor alone, and the MBL-inhibitor complex.
Binding Affinity Prediction:
- The binding energy (ΔE_bind) can be approximated as:
ΔE_bind ≈ E_[MBL:Inhibitor] - (E_[MBL] + E_[Inhibitor]), where each energy is the calculated ground state energy of the respective system. - This quantum-derived energy provides a more reliable starting point for predicting inhibitor potency than classical approximations [57].
- The binding energy (ΔE_bind) can be approximated as:
Diagram 2: MBL inhibitor simulation workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Item / Resource | Function / Description | Relevance to Case Studies |
|---|---|---|
| KRAS G12C Protein (Mutant) | Recombinant protein for in vitro binding and inhibition assays. | Essential for biochemical validation of direct KRAS inhibitors like sotorasib and adagrasib. |
| MBL Enzymes (e.g., NDM-1, VIM-2) | Purified metallo-β-lactamase enzymes for high-throughput screening (HTS). | Used to evaluate the inhibitory activity and potency of novel MBLI candidates. |
| Crystallography Structures (PDB) | Atomic-resolution 3D structures of target proteins (e.g., KRAS, MBLs). | Critical for structure-based drug design, molecular docking, and setting up QM/MM simulations. |
| QM/MM Software Suites | Software (e.g., Terachem, Q-Chem) that enables hybrid quantum-classical simulations. | Provides the framework for partitioning the system and running the MM and QM calculations. |
| Quantum Computing SDKs | Software development kits (e.g., Qiskit, Cirq, TenCirChem) for quantum algorithm implementation. | Used to construct the VQE algorithm, map the Hamiltonian, and interface with quantum hardware/simulators [53]. |
| Classical Optimizers | Algorithms (e.g., COBYLA, SPSA) used in conjunction with VQE. | A classical computing resource that iteratively updates quantum circuit parameters to minimize energy. |
The integration of quantum computing, specifically ground state energy calculations for molecular Hamiltonians, into drug discovery pipelines marks a paradigm shift in tackling previously intractable targets like KRAS and metallo-β-lactamases. While still an emerging field, the development of hybrid quantum-classical workflows for simulating covalent inhibition and metal-ion chelation provides a tangible path toward more rational and accelerated drug design. The ongoing expansion of clinical KRAS inhibitors and the advanced pipeline for MBL inhibitors like taniborbactam validate the biological targets discussed. As quantum hardware continues to scale and algorithms become more refined, the precision and scope of these simulations will only increase, offering a powerful new dimension to the computational scientist's toolkit in the relentless pursuit of novel therapeutics.
Navigating the NISQ Era: Error Mitigation and Advanced Optimization Techniques
For researchers in drug development and quantum chemistry, the accurate calculation of molecular energies is a cornerstone activity. The promise of quantum computing lies in its potential to solve the electronic structure problem—and specifically, to find the ground state energy of molecular Hamiltonians—more efficiently than classical computers. However, the fragile nature of quantum information in today's noisy, intermediate-scale quantum (NISQ) processors poses a significant barrier to realizing this potential [58]. Quantum error mitigation has emerged as a critical discipline, providing a suite of software-based techniques that reduce the impact of noise on computational results without the massive qubit overhead required by full-scale quantum error correction. This guide provides an in-depth examination of these strategies, framed within the practical context of molecular ground state energy estimation, to equip researchers with the knowledge to conduct more reliable quantum simulations.
Foundational Concepts: Noise and Its Impact on Computation
The Nature of Noise in Quantum Processors
In quantum hardware, noise refers to any unwanted interaction that disrupts the ideal evolution of a quantum state. These disruptions originate from various sources, including environmental factors like heat fluctuations, vibrations, and electromagnetic interference, as well as fundamental quantum effects [59] [60]. For superconducting qubits, a prominent architecture for quantum computing, a major source of instability is the interaction between qubits and defect two-level systems (TLS) in the underlying materials. These interactions cause the qubits' relaxation times (T1) to fluctuate dramatically—over 300% on average in some devices—directly impacting the stability and uniformity of processor performance [61]. This noise introduces biases in estimated expectation values, which for quantum chemistry translates directly into inaccurate molecular energy calculations.
Distinguishing Error Correction, Suppression, and Mitigation
It is crucial to distinguish the different approaches to handling errors in quantum computing, as they operate on fundamentally different principles and have varying resource requirements.
Error Suppression: These techniques proactively reduce the likelihood of errors occurring during quantum computation. They operate at the level of quantum control, often integrated directly into the quantum firmware. Methods like dynamic decoupling and Derivative Removal by Adiabatic Gate (DRAG) apply customized control pulses to "deflect" noise or prevent the population of unwanted energy states. Error suppression is deterministic, meaning it reduces errors on every run of a circuit without additional sampling overhead [59] [62].
Error Mitigation: Unlike suppression, error mitigation is a post-processing technique. It does not prevent errors from occurring but instead uses classical post-processing to correct their effect on the computed results, most commonly on estimated expectation values. Techniques like Zero-Noise Extrapolation (ZNE) and Probabilistic Error Cancellation (PEC) accomplish this by running ensembles of slightly modified quantum circuits and combining their results to infer a less noisy, or even noise-free, expectation value [59] [58] [62]. These methods are essential for NISQ computers but come with a significant overhead, often requiring a large number of circuit repetitions.
Quantum Error Correction (QEC): QEC is a algorithmic approach that encodes a single logical qubit of information across many physical qubits. It uses syndrom measurements to detect and correct errors without collapsing the logical quantum state. QEC is the path to fault-tolerant quantum computation but requires a substantial number of high-quality qubits—potentially thousands per logical qubit—a resource threshold not yet met by current hardware [59] [62].
Table: Comparison of Quantum Error Handling Strategies
| Strategy | Operating Principle | Resource Overhead | Hardware Maturity |
|---|---|---|---|
| Error Suppression | Dynamic control to avoid errors | Low (deterministic) | NISQ-ready |
| Error Mitigation | Classical post-processing of noisy results | Moderate to High (sampling) | NISQ-ready |
| Error Correction | Redundant encoding across many physical qubits | Very High (qubit count) | Pre-fault tolerance |
Core Error Mitigation Techniques for Observable Estimation
Accurately estimating the expectation value of a molecular Hamiltonian is the fundamental primitive in ground state energy calculations. The following techniques are central to achieving this on noisy hardware.
Zero-Noise Extrapolation
Core Principle
Zero-Noise Extrapolation (ZNE) operates on a simple but powerful concept: if the effect of noise on an observable can be measured at several known, increased noise strengths, its value in the absence of noise can be inferred through extrapolation [58]. The technique involves intentionally scaling the native noise level of the device, characterized by a strength λ, to a higher level λ' = c * λ (where c > 1).
Methodology
The standard workflow for ZNE is as follows:
- Noise Scaling: A target quantum circuit is executed at its native noise level and at several scaled levels. Common methods for scaling noise include pulse stretching (lengthening gate times) or identity insertion (adding pairs of gates that cancel out logically but increase the circuit's exposure to noise) [58].
- Measurement: For each noise scale factor
c_i, the expectation value of the observable of interest,⟨X(c_i)⟩, is measured. - Extrapolation: A curve is fitted to the data points
(c_i, ⟨X(c_i)⟩). This model is then used to extrapolate the expectation value to the zero-noise limit (c = 0).
Limitations and Considerations
ZNE's major advantage is its generality; it can be applied without a precise knowledge of the underlying noise model. However, its accuracy is highly sensitive to the choice of noise-scaling method, scale factors, and extrapolation model. Errors in the measured expectation values at increased noise levels can be significantly amplified during extrapolation, leading to large uncertainties in the final result [58].
Probabilistic Error Cancellation
Core Principle
Probabilistic Error Cancellation (PEC) is a more advanced technique that relies on a known noise model. It represents an ideal (unitary) quantum gate as a linear combination of noisy, but implementable, operations. By probabilistically sampling from these noisy operations and combining the results with appropriate weights, it is possible to obtain an unbiased estimate of the ideal expectation value [61] [58].
Methodology and Formal Description
The PEC protocol is more involved and requires a pre-characterized device:
- Noise Learning: For each ideal gate
Gin the circuit, a set of noisy implementable operationsΩ = {O_1, ..., O_m}is identified. The ideal gate is then decomposed into a linear combination of these noisy operations:𝒢 = ∑_α η_α 𝒪_α = γ ∑_α P(α) σ(α) 𝒪_α, whereη_αare real coefficients,γ = ∑_α |η_α|is the normalization factor,P(α)=|η_α| / γis a probability distribution, andσ(α)=sign(η_α)[58]. - Circuit Execution:
- For each ideal gate
𝒢_iin the circuit, a noisy operation𝒪_αis sampled from the setΩwith probabilityP_i(α). - This sampled sequence of noisy operations is executed on the quantum processor, producing a final state
ρ_f. - The observable
Xis measured, yielding a resultX_noisy.
- For each ideal gate
- Post-Processing: The ideal expectation value is computed as the sample average
⟨X⟩_ideal = 𝔼 [ γ_tot σ_tot X_noisy ], whereγ_tot = ∏_i γ_iandσ_tot = ∏_i σ_i(α)[58]. The factorγ_totrepresents the sampling overhead.
Limitations and Considerations
PEC can provide an unbiased estimate of the ideal expectation value, but this comes at a cost. The number of samples required to estimate the value with an error δ scales as (2 γ_tot² / δ²) log(2 / ε) [58]. Since γ_tot grows exponentially with the number of gates, the resource overhead can become prohibitive for deep circuits. The accuracy of PEC is also contingent on the precision of the pre-learned noise model.
Stabilizing Noise for Reliable Mitigation
A common requirement for techniques like PEC is an accurate and stable noise model. Recent research highlights that noise instability, particularly from TLS interactions, is a major obstacle. As experimentally demonstrated, this instability can be addressed through active control of the qubit environment.
- Optimized Noise Strategy: This involves actively monitoring the TLS landscape (e.g., by measuring excited state population
P_eas a proxy for T1) and selecting a control parameterk_TLSthat minimizes qubit-TLS interaction before running a computation. This improves worst-case T1 but requires constant monitoring [61]. - Averaged Noise Strategy: This passive approach applies a slow, varying modulation to
k_TLS. This samples many different quasi-static TLS environments across different shots of the experiment, resulting in a more stable average T1 and, consequently, more stable learned noise model parameters for PEC, without the need for constant monitoring [61].
The diagram below illustrates the experimental workflow for stabilizing and characterizing noise to enable high-fidelity error mitigation.
Advanced Frameworks and Experimental Protocols
The Pauli-Lindblad Noise Learning Protocol
For a technique like PEC to be effective, a scalable and accurate noise model is required. The Pauli-Lindblad (SPL) model provides such a framework [61]. The protocol for learning this model is as follows:
- Pauli Twirling: The native noise channel of a gate layer is converted into a Pauli channel by conjugating the gates with random Pauli operators. This ensures the noise can be described purely by Pauli operators.
- Sparse Model Assumption: The noise model
ℰ(ρ) = exp[ℒ](ρ)is constructed from a Lindbladianℒwith Pauli jump termsP_k. The model is sparsified by restricting the set of generators𝒦to one- and two-local Pauli terms that align with the device's qubit connectivity. - Parameter Estimation: The model coefficients
λ_kare characterized by measuring the fidelities of different Pauli operators through a protocol of repeated gate layer applications and measurements. The resultingλ_kparameters provide a snapshot of the noise strength and correlation across the device, which is used to construct the quasi-probability distributions for PEC [61].
Benchmarking for Quantum Chemistry
To fairly evaluate the performance of error-mitigated quantum algorithms against classical methods, structured benchmarking is essential. Frameworks like CSHOREBench and the QB Ground State Energy Estimation Benchmark have been developed for this purpose [63] [64]. These benchmarks assess algorithms on a common set of molecular Hamiltonians and quantum states, accounting for quantum resource utilization (number of measurements) and classical computational runtime. Such benchmarks are vital for identifying the problem domains and scales where quantum methods, enhanced by error mitigation, begin to show a practical advantage.
Table: Key Experimental Findings from Recent Error Mitigation Studies
| Study Focus | Key Quantitative Result | Implication for Ground State Energy Estimation |
|---|---|---|
| TLS Noise Stabilization [61] | T1 fluctuations reduced from >300% to a stable average using averaged noise strategy. | Enables more reliable and repeatable energy calculations by stabilizing the underlying noise model. |
| SPL Noise Model Overhead [61] | Sampling overhead γ = exp(∑_k 2λ_k) tracked over time; stabilization strategies reduced fluctuations in γ. |
Predicts the computational cost (number of samples) for PEC; stability is key for practical resource planning. |
| Decision Diagram Method [63] | Reduced measurements by >80% vs. classical shadows on small molecules. | Lowers the resource burden for estimating the molecular Hamiltonian expectation value. |
The Scientist's Toolkit: Research Reagent Solutions
Beyond abstract algorithms, practical research in this field relies on a suite of software and hardware "reagents." The following table details essential tools for implementing the strategies discussed in this guide.
Table: Essential Tools for Quantum Error Mitigation Research
| Tool / Resource | Type | Primary Function in Error Mitigation |
|---|---|---|
| Mitiq [58] | Software Library | An open-source Python toolkit that provides implementations of ZNE, PEC, and other techniques, interfacing with major quantum SDKs. |
| SPL Model Learning Protocol [61] | Characterization Method | A scalable experimental procedure to learn a sparse noise model for a layer of gates, which is required for performing PEC. |
| QB-GSEE Benchmark [64] | Benchmarking Suite | A curated set of problems for Ground State Energy Estimation, allowing for fair comparison between classical and quantum (error-mitigated) solvers. |
| TLS Control (k_TLS) [61] | Hardware Control | An experimental knob (e.g., a bias electrode) that modulates the qubit-TLS interaction, enabling the stabilization of qubit T1 times. |
Error mitigation is not a silver bullet, but a critical enabler for extracting the maximum possible computational power from today's quantum processors. For researchers focused on molecular ground state energy estimation, techniques like ZNE and PEC, supported by advanced noise characterization and stabilization protocols, are already providing a path toward more accurate Hamiltonian expectation values. The field is rapidly evolving, with progress being driven by tighter integration between hardware control, noise-aware characterization, and robust software tools. By understanding and applying these strategies, scientists can better design quantum experiments, realistically assess their outcomes against classical benchmarks, and push the boundaries of what is possible in computational quantum chemistry and drug discovery.
The accurate simulation of large, biologically relevant molecules stands as a critical challenge in computational chemistry and drug discovery. While quantum computers inherently excel at modeling quantum systems, current hardware remains constrained by limited qubit counts and coherence times. For example, simulating a molecule like insulin would require tracking over 33,000 molecular orbitals, a task far beyond the reach of both classical and quantum computers [40]. This resource gap is particularly pronounced for molecules with strong electron correlation, such as metalloenzymes, where classical methods like density functional theory (DFT) struggle with approximations [13] [65].
Embedding theories have emerged as a powerful strategy to circumvent this limitation. By strategically partitioning a large molecular system into smaller, manageable fragments, these methods allow quantum processors to focus computational resources only on the chemically relevant regions—the subdomains where complex quantum interactions dictate molecular behavior. This approach, which layers multiple embedding techniques, is enabling researchers to run meaningful quantum chemistry calculations on today's noisy intermediate-scale quantum (NISQ) devices, bringing simulations of realistic drug targets and catalysts closer to reality [28] [40].
Core Embedding Theories for Molecular Fragmentation
Embedding methods work by leveraging the locality of chemical phenomena. They partition a system into a region of interest (the fragment) and a surrounding environment, using a different level of theory for each to balance accuracy with computational cost [65].
Table 1: Comparison of Primary Quantum Embedding Theories
| Theory | Partitioning Variable | Key Advantage | Common Use Case |
|---|---|---|---|
| Projection-Based Embedding (PBE) | Orbital Space [28] | Chemically intuitive partitioning; allows different quantum chemistry methods (e.g., CASSCF-in-DFT) [28] [65] | Embedding a high-accuracy active site calculation within a larger molecule treated at a lower level of theory. |
| Density Matrix Embedding Theory (DMET) | Density Matrix [40] [65] | Computationally efficient; provides a rigorous framework for capturing strong electron correlation via Schmidt decomposition [65]. | Strongly correlated systems like transition metal complexes, point defects in solids, and magnetic molecules [65]. |
| Density-Based Embedding (e.g., QM/MM) | Real-Space Density [28] [65] | Allows a quantum mechanical (QM) region to be situated within a classical molecular mechanics (MM) environment of explicit solvent or protein [28]. | Studying enzyme catalysis, solvation effects, and biomolecular systems in a realistic, solvated environment [28]. |
The core principle uniting these methods is the division of labor. A computationally inexpensive method (e.g., DFT or a classical force field) handles the large, chemically inert environment, while a high-accuracy, expensive method (e.g., a quantum algorithm or multireference wavefunction method) is deployed on a small, correlated fragment. The interaction between the fragment and the environment is managed through an embedding potential, which must be carefully constructed to avoid double-counting of correlation energy [65].
Integrated Workflows: A Multi-Scale Approach
No single embedding method is sufficient to bridge the gap from a massive molecular system to a quantum processor. A nested, multi-scale approach is necessary, as visually summarized in the workflow below.
This workflow demonstrates how a large system is progressively distilled for a quantum processing unit (QPU). The process begins with a QM/MM split, situating a target molecule within a vast classical environment [28]. Within the QM region, Projection-Based Embedding (PBE) further partitions the problem, allowing a small, active subsystem to be treated with a high-accuracy quantum solver while the rest of the QM region is handled with a cheaper method like DFT [28]. Finally, qubit subspace techniques exploit molecular symmetries to reduce the qubit overhead of the embedded fragment's Hamiltonian before it is sent to the QPU [28]. The entire process is orchestrated by classical high-performance computing (HPC) resources, exemplifying the hybrid quantum-classical computing paradigm [28] [40].
Experimental Protocols & Key Research Tools
The DMET-SQD Hybrid Protocol
A landmark study demonstrated a practical implementation of this layered approach by combining Density Matrix Embedding Theory (DMET) with the Sample-Based Quantum Diagonalization (SQD) algorithm on an IBM quantum processor [40]. The detailed protocol is as follows:
- System Fragmentation: The target molecule (e.g., a hydrogen ring or cyclohexane) is divided into smaller fragments using DMET.
- Quantum Computation: Each fragment's embedded Hamiltonian is mapped to qubits. The SQD algorithm, known for its noise resilience, is executed on the quantum device (using 27-32 qubits in the cited study).
- Classical Post-Processing: The quantum computer generates samples, which are then processed by a classical computer to solve the Schrödinger equation within a relevant subspace.
- Iteration and Convergence: The DMET algorithm iterates, self-consistently updating the coupling between the fragment and its mean-field environment until the energy and properties converge [40] [65].
This DMET-SQD method successfully computed energy differences between cyclohexane conformers within 1 kcal/mol of classical benchmark results, a threshold considered "chemically accurate" [40].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Computational Tools for Quantum Embedding Experiments
| Tool / "Reagent" | Function | Example/Note |
|---|---|---|
| Embedding Software Framework | Provides the classical infrastructure to partition the molecule and compute the embedding potential. | Tangelo [40] |
| Quantum Algorithm Package | Implements the quantum subroutine (e.g., SQD, VQE) and manages quantum circuit compilation. | Qiskit [40] |
| Error Mitigation Techniques | Reduces the impact of noise on results from NISQ hardware without full error correction. | Zero-Noise Extrapolation (ZNE), Gate Twirling, Dynamical Decoupling [40] [66] |
| Classical Ab Initio Code | Computes the low-level wavefunction or density for the environment and provides molecular integrals. | DFT, Hartree-Fock (HF) codes [40] [65] |
| Hybrid HPC-QPU Platform | The integrated computing infrastructure that coordinates the entire workflow. | e.g., IBM's quantum-centric supercomputing architecture [40] |
Quantitative Landscape: Qubit Requirements for Real-World Targets
The ultimate goal of these methods is to enable the simulation of molecules that are critically important to industry, such as the nitrogen-fixing cofactor FeMoco and the drug-metabolizing enzyme Cytochrome P450. The table below summarizes the estimated qubit resources required for these targets, highlighting how improved hardware and algorithms are reshaping the timeline.
Table 3: Qubit Resource Estimates for Key Industrial Molecules
| Target Molecule | Biological / Industrial Role | Early Qubit Estimate (c. 2021) | Recent Estimate (2025) | Technology / Method Behind Improvement |
|---|---|---|---|---|
| FeMoco (Iron-Molybdenum Cofactor) | Nitrogen fixation in agriculture; model for sustainable fertilizer design [67] [13]. | ~2,700,000 physical qubits [67] [13] | ~99,000 physical qubits [67] | Error-resistant cat qubits (Alice & Bob) [67] |
| Cytochrome P450 | Drug metabolism in humans; crucial for pharmaceutical safety and efficacy [67] [13]. | Similar scale to FeMoco (millions of qubits) [13] | Significant reduction (similar order of magnitude to FeMoco) [67] | Error-resistant cat qubits (Alice & Bob) [67] |
These estimates show a dramatic reduction in physical qubit requirements, largely due to innovations in qubit design that inherently suppress errors. Cat qubits, for instance, protect quantum information through biasing phase-flip errors, which drastically reduces the overhead for error correction [67]. While these numbers are still beyond the scale of today's processors, which typically have 50-1000 physical qubits [25], they illustrate a rapidly closing gap between theoretical resource needs and projected hardware capabilities.
Embedding theories have transformed the qubit count challenge from an insurmountable barrier into a manageable engineering problem. By adopting a multi-scale, fragmented approach, researchers can already use current quantum computers to extract accurate chemical information from subsystems of large molecules, as demonstrated by the DMET-SQD protocol [40]. The continued co-design of hardware, such as error-resilient cat qubits, and software, including more efficient embedding algorithms, is aggressively compressing the timeline to quantum utility in chemistry [15] [67].
The path forward is clear: the integration of quantum embedding workflows into hybrid HPC platforms will be the cornerstone of computational chemistry and drug discovery in the NISQ era and beyond. This strategy allows the quantum processor to focus on what it does best—solving strongly correlated electron problems in small fragments—while leveraging the power of classical computing for everything else. As these methods mature and hardware scales, the routine simulation of currently intractable targets like FeMoco and P450 will move from a distant aspiration to a practical reality, heralding a new era of molecular design [28] [40].
Algorithmic co-design represents a paradigm shift in quantum computing, where hardware capabilities and software solutions are developed in tandem rather than in isolation. Within molecular Hamiltonian ground state searches—a critical task for drug discovery and materials science—this approach tailors quantum algorithms to the specific constraints and strengths of target quantum processors. This whitepaper synthesizes current research to provide a technical guide on co-design methodologies. It covers the theoretical underpinnings of hardware-tailored circuit construction, practical improvements to variational algorithms, and the evaluation of early fault-tolerant protocols. By integrating these strategies, researchers can significantly enhance the performance and feasibility of quantum simulations on today's noisy and tomorrow's fault-tolerant devices.
The pursuit of quantum advantage in molecular science is hampered by the limitations of current quantum hardware. Decoherence, gate errors, and limited qubit connectivity constrain the depth and complexity of executable quantum circuits. Algorithmic co-design directly addresses these challenges by forging a tight feedback loop between the development of quantum hardware, software, and algorithms [68] [69]. The US Department of Energy's Co-Design Center for Quantum Advantage (C2QA), for instance, explicitly focuses on the multiplicative benefits of co-optimizing materials, devices, and software [70]. This methodology moves beyond the conventional model of developing generic algorithms for abstract quantum computers, instead promoting the creation of specialized solutions that achieve greater efficiency on real-world hardware.
In the context of molecular ground state energy estimation—the computational bedrock for understanding chemical properties and reaction dynamics—co-design is particularly impactful. The fundamental goal is to find the lowest eigenvalue of a molecular Hamiltonian, often mapped to a qubit system via transformations like Jordan-Wigner [4] [33]. Co-design principles inform how this problem is approached at every level: from structuring the quantum circuit to minimize hardware-specific overhead, to choosing an ansatz that reflects both molecular physics and device capabilities, and to selecting measurement strategies that reduce resource demands [71] [29]. This integrated approach is poised to accelerate the application of quantum computing to real-world problems in drug discovery and healthcare, where simulating molecular interactions is a key bottleneck [69].
Theoretical Foundations: Hardware-Tailored Circuit Design
A central challenge in near-term quantum algorithms is the simultaneous measurement of commuting Pauli operators, which constitute the molecular Hamiltonian. The efficiency of this measurement step, known as Pauli grouping, directly impacts the number of circuit executions required. Generic diagonalization circuits can lead to an unaffordable SWAP gate overhead on devices with limited hardware connectivity [71]. A co-design approach circumvents this by constructing hardware-tailored (HT) diagonalization circuits.
The theoretical framework for HT circuits leverages the connection between stabilizer states and graph states. Any set of commuting Pauli operators can be simultaneously diagonalized by a Clifford circuit that prepares a stabilizer state. Since every stabilizer state is local-Clifford (LC) equivalent to a graph state, the diagonalization circuit can be decomposed into a layer of single-qubit Clifford gates followed by the uncomputation of a graph state, U_Γ† [71]. The critical co-design insight is to restrict the graph Γ to be a subgraph of the hardware's connectivity graph, Γ_con. This constraint eliminates the need for SWAP gates, ensuring the circuit's two-qubit gate depth is bounded by the maximum degree of the hardware's connectivity graph (e.g., 3 for heavy-hex or 4 for square-lattice architectures) [71].
Table 1: Comparison of Pauli Measurement Strategies
| Strategy | Description | Key Advantage | Key Disadvantage |
|---|---|---|---|
| Tensor Product Basis (TPB) | Diagonalizes sets of qubitwise commuting (QWC) Pauli operators using only single-qubit gates. | Minimal gate depth, simple to implement. | Severely restricts the class of diagonalizable sets, leading to a larger number of required measurements [71]. |
| General Commuting (GC) | Diagonalizes any set of fully commuting Pauli operators. | Drastically reduces the number of measurement circuits required (e.g., to O(n^3) for molecular Hamiltonians) [71]. |
Circuit requires O(n^2) two-qubit gates and may need many SWAP gates on limited-connectivity devices, increasing error and depth [71]. |
| Hardware-Tailored (HT) | Diagonalizes sets of Pauli operators using circuits restricted to the hardware's native connectivity. | Interpolates between TPB and GC; avoids SWAP overhead, making it practical for near-term devices while requiring fewer measurements than TPB [71]. | Requires a more complex classical pre-processing step to group Paulis into jointly-HT-diagonalizable sets [71]. |
This framework provides a flexible foundation for optimizing Hamiltonian measurement. By tailoring the diagonalization circuit to the hardware, it achieves a favorable trade-off between the number of circuit executions and the noise resilience of each individual circuit.
Algorithmic Innovations for Near-Term Hardware
Adaptive Ansätze with Hardware Awareness
The Variational Quantum Eigensolver (VQE) is a leading near-term algorithm for ground state energy estimation. Its performance heavily depends on the choice of ansatz. Adaptive variants like ADAPT-VQE build a problem-specific ansatz by iteratively adding operators from a predefined pool based on their predicted contribution to energy lowering [4]. A co-design extension of this concept, K-ADAPT-VQE, improves efficiency by adding K operators in "chunks" at each iteration, thereby reducing the total number of costly classical optimization loops and quantum function evaluations [4].
The operator pool itself can be co-designed. While a common approach uses all symmetry-allowed single and double excitations from a Unitary Coupled-Cluster (UCCSD) ansatz, the physical implementation of these operators must be considered. The resulting quantum circuits must be compiled down to the native gate set and connectivity of the target device. An awareness of this process can inform the construction of the operator pool or the selection metric in ADAPT-VQE, potentially favoring operators that result in shower, more hardware-native circuits.
Generative Quantum Eigensolver
Moving beyond fixed or adaptively built parameterized circuits, the Generative Quantum Eigensolver (GQE) represents a radical co-design approach. In GQE, a classical generative model (e.g., a transformer neural network) is trained to generate entire quantum circuits [72]. Unlike VQE, no parameters are embedded in the quantum circuit itself; all parameters reside in the classical model, which is optimized to output circuits that minimize the energy of a given Hamiltonian.
The conditional-GQE variant uses an encoder-decoder transformer architecture to generate context-aware circuits [72]. For a molecular problem, the Hamiltonian's characteristics (e.g., its Pauli coefficients) can be encoded as a graph and fed into the model, which then generates a tailored quantum circuit. This method separates the classical learning from the quantum execution, potentially overcoming expressibility limitations of traditional parameterized circuits and directly incorporating hardware constraints into the generative process [72].
Experimental Protocols and Methodologies
Protocol: Hardware-Tailored Pauli Grouping
This protocol details the steps for grouping Hamiltonian Pauli terms into sets measurable using hardware-tailored diagonalization circuits [71].
Hamiltonian Preparation: Generate the qubit Hamiltonian
Hfor the target molecule. Using quantum chemistry packages likePySCF[4], compute the one- and two-electron integrals. Then, using a tool likeOpenFermion[4], perform a fermion-to-qubit mapping (e.g., Jordan-Wigner) to expressHas a linear combination of Pauli terms:H = Σ_i c_i P_i.Stabilizer Group Synthesis: For a candidate set of commuting Pauli operators
{P_1, ..., P_m}, find a stabilizer groupSthat contains them. This may involve replacing someP_jwith-P_jto ensure-I⊗nis not in the group.Graph State Identification: Find a graph
Γand a layer of single-qubit Clifford gatesU_1, ..., U_nsuch that the stabilizer state|ψ_S⟩is LC-equivalent to the graph state|Γ⟩. That is,(U_1 ⊗ ... ⊗ U_n)|ψ_S⟩ = |Γ⟩.Hardware Constraint Integration: Check if the graph
Γis a subgraph of the hardware connectivity graphΓ_con. If true, the diagonalization circuitU = (U_1^† ⊗ ... ⊗ U_n^†) * U_Γ^†is hardware-tailored and requires no SWAP gates. The circuitU_Γ^†is built by reversing the graph state preparation circuit, which consists of a layer of Hadamard gates followed by CZ gates for every edge inΓ[71].Grouping Optimization: Implement a classical algorithm to partition all Pauli terms of the Hamiltonian into a minimal number of jointly-HT-diagonalizable sets, following the above procedure.
Protocol: K-ADAPT-VQE for Molecular Ground States
This protocol describes running the K-ADAPT-VQE algorithm to find a molecular ground state energy [4].
Initialization:
- Prepare the molecular Hamiltonian
Has in Step 1 of the previous protocol. - Prepare the Hartree-Fock (HF) state
|Ψ_HF⟩as the initial reference state. - Define an operator pool
A, typically composed of fermionic excitation operators (e.g., from UCCSD) mapped to qubit operators. - Initialize the ansatz circuit
V(θ)to be empty, or to contain the HF state preparation.
- Prepare the molecular Hamiltonian
Iterative Ansatz Construction:
- Gradient Calculation: For each operator
τ_iin the poolA, calculate the energy gradient∂E/∂θ_i = ⟨Ψ|[H, τ_i]|Ψ⟩, where|Ψ⟩is the current ansatz state. This requires evaluating the expectation value on a quantum computer. - Operator Selection: Instead of selecting the single operator with the largest gradient magnitude, select the top
Koperators{τ_1, ..., τ_K}. - Ansatz Expansion: Append the selected operators to the ansatz:
V(θ) → V(θ) * [exp(θ_{n+1} τ_1) * ... * exp(θ_{n+K} τ_K)]. Initialize the new parametersθ_{n+1}, ..., θ_{n+K}to zero. - Circuit Compilation: Compile the updated ansatz
V(θ)into the native gates and topology of the target quantum hardware.
- Gradient Calculation: For each operator
Parameter Optimization:
- Use a classical optimizer (e.g., COBYLA, CMA-ES) to minimize the energy
E(θ) = ⟨0| V†(θ) H V(θ) |0⟩. - The quantum computer is used to evaluate the cost function
E(θ)for different parameter valuesθ.
- Use a classical optimizer (e.g., COBYLA, CMA-ES) to minimize the energy
Convergence Check: Repeat the iterative process from Step 2 until the energy change falls below a predefined chemical accuracy threshold (e.g., 1.6 mHa).
Evaluation and Resource Analysis in the Early Fault-Tolerant Era
As quantum computing progresses towards fault tolerance, co-design principles remain essential for managing resource overhead. A promising alternative to the demanding Quantum Phase Estimation (QPE) algorithm is the Cumulative Distribution Function (CDF) method. This approach identifies the ground state energy by locating the first discontinuity in the CDF of the Hamiltonian's spectral measure with respect to the initial state [29].
A key co-design challenge here is preparing an initial state with sufficient overlap with the true ground state. A signal processing-based co-design approach has been proposed to automatically extract energy estimates even when the exact overlap is unknown. This method refines classical estimates by targeting the low-energy support of the initial state, requiring several orders of magnitude fewer samples than theoretical upper bounds suggest [29]. For example, numerical experiments on a 26-qubit fully connected Heisenberg model using a low-bond-dimension Density Matrix Renormalization Group (DMRG) initial state showed that quantum predictions aligned closely with fully converged DMRG energies while being far more resource-efficient than QPE [29].
Table 2: Resource Comparison for Ground State Energy Algorithms
| Algorithm | Key Principle | Circuit Depth | Sample Complexity | Suitability |
|---|---|---|---|---|
| VQE | Variational principle; hybrid quantum-classical optimization. | Shallow, but many iterations required. | High (due to many iterations and measurements). | Noisy Intermediate-Scale Quantum (NISQ) devices [33]. |
| QPE | Quantum phase estimation; projects energy eigenstate. | Very deep, requires long coherence. | Theoretically lower, but requires full fault tolerance. | Large-scale fault-tolerant quantum computers. |
| CDF Method | Signal processing of the Hamiltonian's spectral CDF. | Moderate, shorter than QPE. | Fewer samples than QPE in practice; constant factor improvements reported [29]. | Early Fault-Tolerant Quantum Computers [29]. |
This analysis demonstrates that co-design extends to choosing the core algorithmic primitive based on an honest assessment of the available hardware's capabilities and error rates, optimizing for the overall resource cost of a scientifically meaningful result.
The Scientist's Toolkit: Essential Research Reagents
This section details the key software, hardware, and methodological "reagents" required to implement the co-designed strategies discussed in this whitepaper.
Table 3: Essential Tools for Quantum Algorithm Co-Design Research
| Tool / Reagent | Type | Primary Function | Relevance to Co-Design |
|---|---|---|---|
| PySCF [4] | Software Library | Performs classical quantum chemistry calculations (e.g., Hartree-Fock, integral computation). | Generates the molecular Hamiltonian, the target problem for quantum simulation. |
| OpenFermion [4] | Software Library | Translates electronic structure Hamiltonians from fermionic to qubit representations. | Provides the bridge between quantum chemistry problems and quantum circuit models, supporting various mappings (JW, BKT). |
| Graph State Framework [71] | Theoretical Framework | Represents quantum states via graphs, with edges defining entanglement via CZ gates. | Enables the construction of hardware-tailored diagonalization circuits by matching the graph to device connectivity. |
| PennyLane [33] | Quantum Software Framework | A cross-platform library for differentiable programming of quantum computers. | Used to define, execute, and optimize variational algorithms (like VQE); supports various devices and simulators. |
| Conditional-GQE Model [72] | Machine Learning Model | An encoder-decoder transformer trained to generate context-aware quantum circuits. | Embodies co-design by using a classical AI to generate hardware-efficient circuits specific to a problem Hamiltonian. |
| Hardware Connectivity Graph (e.g., Heavy-hex) [71] | Hardware Specification | The physical topology of qubits and their usable couplings on a quantum processor. | The central constraint around which circuits (e.g., for measurement or ansatz) are tailored to minimize SWAP overhead. |
Algorithmic co-design is not a luxury but a necessity for unlocking the potential of quantum computing in the molecular sciences. By tailoring diagonalization circuits to hardware connectivity, adapting ansatz construction to minimize resource use, and selecting algorithms suited for the early fault-tolerant era, researchers can significantly extend the computational reach of both current and future quantum processors. The integration of advanced classical methods, from graph theory to machine learning, with quantum execution creates a powerful symbiotic relationship. As the field matures, co-design methodologies will become increasingly sophisticated, driven by close collaboration between quantum chemists, algorithm developers, and hardware engineers. This collaborative, application-focused approach is the most promising path toward achieving quantum advantage in simulating molecular systems and accelerating critical research in drug development and materials discovery.
The pursuit of accurately solving molecular Hamiltonians to determine ground-state energies represents a core challenge in computational chemistry and drug discovery. While quantum computers hold immense promise for this task, current hardware operates in the noisy intermediate-scale quantum (NISQ) era, characterized by significant limitations in qubit coherence, gate fidelity, and qubit count. These constraints have catalyzed the development of sophisticated hybrid methodologies that strategically combine quantum processing with classical computational resources. This technical guide examines the cutting-edge integration of advanced sampling techniques and classical post-processing algorithms that are extending the capabilities of quantum computations for molecular systems. By framing these approaches within the quantum-centric supercomputing (QCSC) paradigm—where quantum processors handle specific subroutines while classical systems manage preprocessing, error mitigation, and post-processing—researchers can achieve unprecedented accuracy in molecular simulations, moving closer to the coveted goal of chemical accuracy (±1 kcal/mol) for biologically relevant systems [40] [44].
Theoretical Foundations
The Sampling-Post-Processing Paradigm
The fundamental insight driving recent advancements is that raw quantum computational output often contains valuable signal obscured by hardware noise and statistical uncertainty. Rather than treating quantum processors as standalone solutions, the sampling-post-processing paradigm recognizes them as specialized components within a larger computational workflow. In this framework, quantum computers generate samples from parametrized quantum circuits that approximate molecular wavefunctions, while classical algorithms post-process these samples to extract accurate energy estimates [73] [74]. This division of labor leverages the quantum computer's ability to efficiently explore exponentially large Hilbert spaces while utilizing classical resources for error mitigation, symmetry enforcement, and high-precision diagonalization.
The theoretical justification for this approach rests on several key principles. First, the variational principle ensures that energy estimates derived from post-processed quantum states provide upper bounds to the true ground-state energy. Second, many molecular systems possess known symmetries (particle number, spin, etc.) that can be enforced during classical post-processing to project out unphysical components introduced by hardware noise. Third, the statistical distribution of quantum measurements contains information about dominant electronic configurations that can be intelligently leveraged to construct compact yet accurate classical representations of the quantum state [44] [75].
Key Algorithmic Frameworks
Several algorithmic frameworks have emerged as particularly effective for the sampling-post-processing paradigm:
Sample-Based Quantum Diagonalization (SQD): This method uses a quantum device to sample electronic configurations from an approximate wavefunction, then employs classical high-performance computing (HPC) resources to post-process these samples against known symmetries. The recovered configurations form a subspace in which the Schrödinger equation is solved classically [40] [44].
Density Matrix Embedding Theory (DMET) with SQD: DMET partitions molecular systems into smaller fragments embedded in an approximate environment, with SQD handling the quantum simulation of these fragments. This combination allows for the simulation of chemically relevant molecules using only 27-32 qubits while maintaining accuracy within 1 kcal/mol of classical benchmarks [40].
Hybrid Quantum-Neural Wavefunctions: Approaches like the pUNN (paired Unitary Coupled-Cluster with Neural Networks) framework combine efficient quantum circuits with neural networks. The quantum circuit learns the quantum phase structure, while the neural network corrects the amplitude, achieving near-chemical accuracy with enhanced noise resilience [75].
Advanced Sampling Methodologies
Configuration Sampling with Ansatz Optimization
The efficacy of classical post-processing depends critically on the quality of samples generated by quantum circuits. Advanced sampling begins with carefully constructed ansatzes that balance expressibility with hardware feasibility:
Table 1: Comparison of Quantum Ansatzes for Molecular Ground State Sampling
| Ansatz Type | Qubit Requirements | Circuit Depth | Key Advantages | Sampling Limitations |
|---|---|---|---|---|
| Unitary Coupled Cluster (UCCSD) | 2N qubits for N orbitals | High (exponential scaling) | Physically motivated, exact in limit | Impractical on current devices |
| Local UCC (LUCJ) | 2N qubits for N orbitals | Moderate (polynomial scaling) | Approximates UCCSD with reduced depth | May miss long-range correlations |
| Hardware-Efficient | N qubits | Low (linear scaling) | Implementable on current hardware | Lacks physical constraints |
| pUCCD with Neural Augmentation | N qubits (plus classical NN) | Low (linear scaling) | Seniority-zero subspace with neural correction | Requires classical co-processing |
For the UCCSD ansatz, the wavefunction is constructed as ( |\Psi\rangle = e^{T - T^\dagger} |\Phi0\rangle ), where ( T ) generates single and double excitations from the reference state ( |\Phi0\rangle ). While theoretically optimal, this approach requires circuit depths prohibitive on current hardware. The LUCJ ansatz addresses this limitation by restricting excitations to local clusters, dramatically reducing circuit depth while preserving much of the expressive power. For instance, LUCJ simulations of methane dimer (16 electrons, 24 orbitals) achieved execution times of approximately 229 seconds on IBM's ibm_kyiv device, making chemically relevant simulations feasible [44].
Practical Implementation Protocols
Implementing advanced sampling for molecular systems requires careful workflow design:
Active Space Selection: Identify chemically relevant orbitals using automated tools like the Active Space Transformer in Qiskit or the AVAS (Automated Valence Active Space) method. For aluminum clusters (Al⁻ to Al₃⁻), a 3-orbital active space with 4 electrons has proven effective [76] [77].
Ansatz Parameter Optimization: Employ hybrid quantum-classical optimization loops where quantum circuits prepare trial states and classical optimizers (e.g., SLSQP, COBYLA) adjust parameters to minimize energy. The Variational Quantum Eigensolver (VQE) framework typically requires 100-500 iterations for convergence [76].
Circuit Execution Strategy: Distribute sampling across multiple quantum processor units (QPUs) to collect sufficient statistics. For the DMET-SQD approach applied to cyclohexane conformers, 8,000-10,000 samples per fragment were necessary to achieve chemical accuracy [40].
The diagram below illustrates the complete advanced sampling workflow:
Figure 1: Advanced Sampling Workflow for Molecular Systems. This diagram illustrates the iterative hybrid quantum-classical procedure for generating configuration samples from parametrized quantum circuits.
Classical Post-Processing Techniques
Sample-Based Quantum Diagonalization (SQD)
SQD represents one of the most powerful post-processing techniques for extracting accurate energies from noisy quantum samples. The methodology proceeds through several well-defined stages:
Configuration Recovery: Raw quantum measurements often violate known physical symmetries due to hardware noise. The S-CORE (Self-Consistent Configuration Recovery) algorithm processes these samples to identify and repair corrupted configurations, enforcing conservation of particle number and spin symmetry [44].
Subspace Construction: The recovered configurations form a many-body subspace ( \mathcal{S} = { |C1\rangle, |C2\rangle, \dots, |C_M\rangle } ) where ( M ) is typically ( 10^7 - 10^9 ) for molecular systems with 16-54 qubits [44].
Diagonalization: The molecular Hamiltonian is projected into this subspace ( H{ij} = \langle Ci|H|C_j\rangle ), and the resulting generalized eigenvalue problem is solved using distributed classical HPC resources.
The exceptional efficacy of SQD is demonstrated by its application to the methane dimer system (54-qubit circuit), where it successfully diagonalized a subspace of 2.49×10⁸ configurations, producing binding energies within 1 kcal/mol of CCSD(T) benchmarks [44].
Error Mitigation through Classical Processing
Beyond SQD, several specialized techniques enhance the signal-to-noise ratio in quantum computations:
Algorithmic Fault Tolerance: Techniques like those implemented in QuEra's published approaches reduce quantum error correction overhead by up to 100 times, moving timelines for practical quantum computing substantially forward [15].
Noise-Aware Post-Selection: By filtering quantum measurements based on physical constraints (particle number, spin symmetry), researchers can significantly improve accuracy even without knowledge of the specific noise model.
Zero-Noise Extrapolation: This technique runs the same quantum circuit at multiple noise levels (achieved through circuit folding or pulse stretching) and extrapolates to the zero-noise limit.
Table 2: Performance Metrics of Post-Processing Techniques on Molecular Systems
| Molecular System | Qubit Count | Method | Classical Benchmark | Energy Deviation | Key Post-Processing |
|---|---|---|---|---|---|
| Water Dimer | 27 | SQD | CCSD(T) | < 1.000 kcal/mol | Configuration recovery & subspace diagonalization |
| Methane Dimer | 36 | SQD | CCSD(T) | < 1.000 kcal/mol | Subspace construction (2.49×10⁸ configurations) |
| Cyclohexane Conformers | 32 | DMET-SQD | HCI | < 1.000 kcal/mol | Fragment embedding & error mitigation |
| Aluminum Clusters | Varies | VQE with noise models | CCCBDB | < 0.2% error | Noise model simulation & parameter optimization |
Hybrid Quantum-Neural Processing
The pUNN framework represents a particularly innovative approach to post-processing that integrates neural networks directly with quantum computations. In this methodology:
A quantum circuit (typically pUCCD with linear depth) generates the core wavefunction in the seniority-zero subspace.
A neural network operator ( \mathcal{M} ) acts as a non-unitary post-processing transformation: ( |\Psi\rangle = \mathcal{M} \hat{E}(|\psi\rangle \otimes |0\rangle) ), where ( \hat{E} ) is an entanglement circuit [75].
The neural network architecture consists of L dense layers with ReLU activations, where the number of parameters scales as ( K^2N^3 ) for N qubits, providing substantial expressive power while remaining computationally tractable.
This approach demonstrated remarkable noise resilience when implemented on superconducting quantum hardware for the isomerization reaction of cyclobutadiene, a challenging multi-reference system [75].
Experimental Protocols and Validation
Standardized Benchmarking Methodology
To ensure rigorous validation of sampling and post-processing techniques, researchers should implement comprehensive benchmarking protocols:
System Selection: Choose molecular systems with well-characterized electronic structure properties. The aluminum clusters (Al⁻, Al₂, Al₃⁻) provide intermediate complexity with reliable classical benchmarks from CCCBDB [76] [77]. For non-covalent interactions, water and methane dimers serve as excellent test cases [44].
Parameter Variation: Systematically explore key algorithmic parameters including:
- Classical optimizers (SLSQP, COBYLA, L-BFGS-B)
- Circuit types (UCCSD, LUCJ, hardware-efficient)
- Basis sets (STO-3G, 6-31G, cc-pVDZ)
- Number of circuit repetitions/reps
- Quantum simulator types (statevector, shot-based, noisy)
Noise Modeling: Incorporate realistic noise models (such as those from IBM) to evaluate performance under conditions mimicking actual hardware. Research shows that with proper post-processing, VQE can maintain errors below 0.2% even with simulated noise [76].
Validation Metrics and Accuracy Standards
Chemical accuracy (1 kcal/mol or 1.6 mHa) serves as the gold standard for molecular energy calculations. Beyond absolute energy accuracy, researchers should evaluate:
Relative Energies: Differences between molecular conformers (e.g., cyclohexane chair, boat, twist-boat) often provide more chemically relevant validation than total energies [40].
Potential Energy Surfaces: Scanning along reaction coordinates or intermolecular distances tests the robustness of methods across different electronic configurations.
Resource Scaling: Tracking computational time, qubit requirements, and circuit depth as function of system size provides practical guidance for future applications.
The following workflow diagram illustrates the complete validation process for post-processing techniques:
Figure 2: Validation Workflow for Post-Processing Techniques. This process ensures that classical post-processing methods reliably extract chemically accurate energies from noisy quantum samples.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of advanced sampling and post-processing requires familiarity with both quantum and classical software frameworks:
Table 3: Essential Tools for Quantum Computational Chemistry Research
| Tool/Framework | Primary Function | Application in Sampling/Post-Processing | Access Method |
|---|---|---|---|
| Qiskit Nature | Quantum computational chemistry | Active space selection, ansatz construction, VQE implementation | Open source (Python) |
| Tangelo | Classical computational chemistry | DMET implementation, fragment-based simulations | Open source (Python) |
| PySCF | Electronic structure theory | Hamiltonian generation, classical benchmarks | Open source (Python) |
| Amazon Braket | Quantum computing service | Hybrid job execution, multi-device sampling | Cloud platform |
| Q-CTRL Fire Opal | Quantum error suppression | Circuit optimization, error mitigation | Cloud platform |
| BenchQC | Benchmarking toolkit | Performance evaluation, parameter optimization | Open source (Python) |
| JARVIS-DFT | Materials database | Structure retrieval, benchmark comparison | Web database |
The integration of advanced sampling techniques with sophisticated classical post-processing represents a pivotal advancement in quantum computational chemistry. By leveraging hybrid frameworks such as SQD, DMET, and quantum-neural architectures, researchers can now extract chemically accurate energies from noisy quantum devices of modest scale. These approaches effectively distribute the computational burden, using quantum processors for tasks where they provide natural advantages while employing classical resources for error mitigation, symmetry enforcement, and high-precision linear algebra. As quantum hardware continues to evolve with improving qubit counts and error rates—exemplified by IBM's roadmap targeting 1,000+ qubit systems and error rates below 0.000015%—the sampling-post-processing paradigm will likely remain essential for bridging the gap between current capabilities and the future promise of fault-tolerant quantum computation for molecular systems. For researchers in pharmaceutical development and materials science, these methodologies offer a practical pathway to leverage quantum computing for challenging electronic structure problems today while building foundation for more substantial advantages in the coming years.
Benchmarking Quantum Accuracy: Verifiable Advantage and Performance Analysis
The pursuit of chemical accuracy, defined as an error margin of 1.6 × 10−3 Hartree in energy calculations, represents a critical milestone for quantum computing applications in molecular simulation [78]. This precision threshold is not merely symbolic; it determines the practical utility of computational methods for predicting reaction rates, binding affinities, and other chemically significant properties where errors exceeding 1 kcal/mol can lead to erroneous conclusions in drug design pipelines [79]. For quantum computing to demonstrate genuine advantage in molecular Hamiltonian ground state search, it must reliably achieve this accuracy while surpassing the capabilities of established classical computational chemistry methods.
The coupled cluster singles, doubles, and perturbative triples (CCSD(T)) method has long been regarded as the "gold standard" in quantum chemistry, providing benchmark accuracy for molecular systems [80]. However, its computational cost scales as O(N7) with system size, creating an exponential bottleneck that limits applications to larger molecules and complex systems such as enzymes with transition metal centers [81]. This scaling problem has motivated the exploration of quantum algorithms as a potential pathway to maintain CCSD(T)-level accuracy with more favorable scaling. Recent research has begun establishing "platinum standards" through agreement between complementary high-level methods like CCSD(T) and quantum Monte Carlo, further refining accuracy targets for quantum algorithms [79].
This technical guide examines the current landscape of benchmarking quantum computational results against CCSD(T) and other classical methods, providing researchers with experimental protocols, performance comparisons, and implementation frameworks to rigorously evaluate progress toward chemical accuracy in quantum algorithms for molecular ground state energy calculation.
Classical Benchmarks: Understanding the Quantum Chemistry Landscape
Hierarchy of Classical Methods
Classical computational chemistry methods form a hierarchy of increasing accuracy and computational cost, providing the essential benchmarking foundation for evaluating quantum algorithm performance. Understanding this landscape is crucial for meaningful comparisons.
Table 1: Classical Computational Chemistry Methods for Ground State Energy Calculation
| Method | Time Complexity | Key Characteristics | Limitations |
|---|---|---|---|
| Hartree-Fock (HF) | O(N⁴) | Mean-field approximation, computationally efficient | Lacks electron correlation [80] |
| Density Functional Theory (DFT) | O(N³) | Balances efficiency and accuracy, various functionals | Exchange-correlation functional approximation [81] |
| Møller-Plesset 2nd Order (MP2) | O(N⁵) | Includes electron correlation via perturbation theory | Can overestimate correlation energy |
| Coupled Cluster Singles/Doubles (CCSD) | O(N⁶) | High accuracy for single-reference systems | High computational cost [81] |
| CCSD(T) | O(N⁷) | "Gold standard," includes perturbative triples | Prohibitive for large systems [81] |
| Full Configuration Interaction (FCI) | O*(4ⁿ) | Exact solution within basis set | Computationally prohibitive [80] |
Accuracy Expectations and Limitations
Each classical method has characteristic accuracy ranges, with CCSD(T) typically achieving errors below 1 kcal/mol for systems where it is applicable [79]. However, for systems with strong correlation effects or multireference character, such as the stretched H4 system or transition metal complexes, even CCSD(T) can struggle, creating opportunities for quantum advantage [9]. The recent development of the QUID (QUantum Interacting Dimer) benchmark framework, which establishes robust binding energies through complementary CCSD(T) and quantum Monte Carlo calculations, highlights the ongoing evolution of accuracy standards in the field [79].
Quantum Algorithms for Ground State Energy Calculation
Variational Quantum Eigensolver (VQE)
The Variational Quantum Eigensolver (VQE) has emerged as a leading near-term quantum algorithm for molecular ground state energy calculation, employing a hybrid quantum-classical approach [80]. VQE operates on the variational principle, using parameterized quantum circuits (ansatzes) to prepare trial wavefunctions and classical optimizers to minimize the expectation value of the Hamiltonian:
E(θ) = ⟨ψ(θ)|Ĥ|ψ(θ)⟩ ≥ E₀
where E(θ) is the energy of the trial state |ψ(θ)⟩, Ĥ is the system Hamiltonian, θ represents the circuit parameters, and E₀ is the exact ground-state energy [80].
The performance of VQE depends critically on three components: ansatz selection, parameter initialization, and classical optimizer choice [80]. Common ansatzes include:
- UCCSD: Chemically inspired, strong performance but deep circuits
- k-UpCCGSD: Shallower circuits, suitable for noisy hardware
- Hardware-efficient: Minimal depth but limited chemical intuition
Research on the silicon atom ground state demonstrates that parameter initialization plays a decisive role in algorithm stability, with zero initialization often providing superior convergence [80]. For optimizers, adaptive methods like ADAM combined with chemically inspired ansatzes typically yield the best performance.
Quantum Phase Estimation (QPE) and Dissipative Approaches
Beyond VQE, other quantum algorithms show promise for ground state energy calculation. Quantum Phase Estimation (QPE) provides a direct route to energy eigenvalues without the optimization challenges of VQE but requires greater circuit depth and coherence times, making it suitable for fault-tolerant systems [9]. Recent analysis suggests QPE could surpass high-accuracy classical methods like FCI and CCSD(T) within the coming decade, with projected advantage timelines around 2031-2032 for FCI and 2034-2036 for CCSD(T) [81].
A novel approach using dissipative engineering through Lindblad dynamics offers a parameter-free alternative to variational methods [9]. This method designs jump operators that systematically shift population toward the ground state, proving effective for systems like BeH₂, H₂O, and Cl₂, even in strongly correlated regimes where traditional methods struggle [9]. The approach introduces Type-I and Type-II jump operators, with Type-II preserving particle number symmetry for more efficient simulation in the full configuration interaction space [9].
Benchmarking Methodologies and Performance Comparisons
Quantitative Performance Projections
Recent comprehensive analyses project timelines for quantum advantage across different computational chemistry methods, accounting for anticipated improvements in quantum hardware and algorithms.
Table 2: Projected Quantum Advantage Timeline for Quantum Phase Estimation vs. Classical Methods
| Classical Method | Time Complexity | QPE Advantage Projection | Key Factors |
|---|---|---|---|
| Density Functional Theory (DFT) | O(N³) | >2050 | QPE complexity O(N²/ε) insufficient [81] |
| Hartree Fock (HF) | O(N⁴) | ~2044 | Hardware improvements needed [81] |
| MP2 | O(N⁵) | ~2038 | Moderate quantum speedup [81] |
| CCSD | O(N⁶) | ~2036 | Favorable scaling comparison [81] |
| CCSD(T) | O(N⁷) | ~2034 | Significant scaling advantage [81] |
| FCI | O*(4ⁿ) | ~2031-2032 | Exponential vs. polynomial scaling [81] |
Note: Projections assume ε = 10⁻³ accuracy and favorable hardware development.
Experimental Protocols for Rigorous Benchmarking
VQE Configuration Testing Protocol
To systematically evaluate VQE performance against classical benchmarks:
- System Selection: Begin with small systems (H₂, LiH) where CCSD(T) and FCI references are computationally feasible, then progress to medium-sized atoms (e.g., silicon) and molecules with known experimental data [80].
- Hamiltonian Preparation: Transform the electronic structure Hamiltonian to qubit representation using Jordan-Wigner or Bravyi-Kitaev mappings [80].
- Ansatz Comparison: Implement multiple ansatzes (UCCSD, k-UpCCGSD, hardware-efficient) with identical initial conditions [80].
- Optimizer Benchmarking: Test various optimizers (gradient descent, SPSA, ADAM) with controlled parameter initialization strategies [80].
- Error Mitigation: Apply readout error mitigation, zero-noise extrapolation, and other techniques to enhance accuracy [78].
- Precision Assessment: Compare final energies against CCSD(T) references, calculating both absolute error and achievement of chemical accuracy (1.6 × 10−3 Hartree) [78].
Dissipative Approach Implementation
For implementing dissipative ground state preparation:
- Jump Operator Selection: Choose Type-I (particle-number non-conserving) or Type-II (particle-number conserving) jump operators based on system requirements [9].
- Lindbladian Simulation: Employ Monte Carlo trajectory-based algorithms for simulating Lindblad dynamics [9].
- Mixing Time Analysis: Measure time required to reach the target steady state to within chemical accuracy [9].
- Active Space Strategy: Reduce jump operator count while preserving convergence behavior for computational efficiency [9].
Practical Implementation and Error Mitigation
Measurement Techniques and Precision Enhancement
Achieving chemical precision on near-term quantum hardware requires sophisticated measurement strategies:
- Locally Biased Random Measurements: Reduce shot overhead by prioritizing measurement settings with greater impact on energy estimation [78].
- Quantum Detector Tomography (QDT): Mitigate readout errors by characterizing noisy measurement effects and building unbiased estimators [78].
- Blended Scheduling: Address time-dependent noise by interleaving circuits for different Hamiltonians with QDT circuits [78].
- Informationally Complete (IC) Measurements: Enable estimation of multiple observables from the same dataset, crucial for measurement-intensive algorithms like ADAPT-VQE [78].
Recent demonstrations on IBM quantum hardware have reduced measurement errors from 1-5% to 0.16%, approaching chemical precision despite readout errors on the order of 10⁻² [78].
The Researcher's Toolkit: Essential Components for Quantum Chemistry Benchmarking
Table 3: Essential Research Tools for Quantum-Chemistry Benchmarking
| Tool/Component | Function | Implementation Examples |
|---|---|---|
| Molecular Hamiltonians | Define electronic structure problem | H₂, LiH, BeH₂, H₂O, Si atom, BODIPY [80] [78] |
| Ansatz Circuits | Parameterized wavefunction preparation | UCCSD, k-UpCCGSD, PUCCD, hardware-efficient [80] |
| Classical Optimizers | Parameter optimization | ADAM, SPSA, gradient descent [80] |
| Error Mitigation | Counteract hardware noise | Zero-noise extrapolation, readout error mitigation [78] |
| Measurement Protocols | Efficient observable estimation | Locally biased classical shadows, informationally complete measurements [78] |
| Reference Calculations | Provide benchmark accuracy | CCSD(T), FCI, quantum Monte Carlo [79] |
The rigorous benchmarking of quantum computing results against CCSD(T) and other classical methods reveals a field in rapid transition. Current hybrid quantum-classical approaches, particularly VQE with advanced error mitigation, are beginning to approach chemical accuracy for small molecular systems, while dissipative methods offer promising parameter-free alternatives [80] [9] [78]. Projections suggest that quantum advantage for highly accurate simulations may emerge within the next decade, particularly for systems where classical methods face fundamental limitations [81].
The path toward universal quantum advantage in computational chemistry will likely proceed through distinct phases: initial demonstrations on specific problem classes with known limitations in classical methods, followed by broader applicability as hardware improves and algorithms mature. For researchers in this field, maintaining rigorous benchmarking practices against established classical methods remains essential for distinguishing genuine progress from speculative claims. As the field advances, the integration of quantum simulations into broader computational workflows—such as drug discovery pipelines and materials design—will provide the ultimate test of whether quantum algorithms can deliver on their promise to revolutionize molecular simulation.
The pursuit of quantum advantage has entered a new era, moving beyond abstract computational supremacy to verifiable, scientifically relevant tasks. This whitepaper examines the groundbreaking demonstration of verifiable quantum advantage achieved through the measurement of Out-of-Time-Order Correlators (OTOCs) using the Quantum Echoes algorithm. We detail how this approach, implemented on Google's Willow quantum processor, enables tasks that are provably beyond classical simulation while producing physically meaningful, verifiable results. The core principles, experimental methodologies, and implications for molecular Hamiltonian ground state search are explored, providing researchers with a comprehensive technical overview of this transformative development.
The field of quantum computing has reached a critical inflection point. While previous demonstrations established the raw computational potential of quantum devices, the quest for practical utility has been hampered by two significant challenges: the non-verifiability of results and limited practical applicability [82]. Early benchmarks, such as random circuit sampling, demonstrated that quantum processors could perform tasks infeasible for classical computers but offered limited scientific utility as the same bitstring never appears twice in a large quantum system [83].
The concept of verifiable quantum advantage represents a paradigm shift. It describes a quantum computation that is not only beyond classical reach but also produces a verifiable outcome—a result that can be confirmed by another quantum computer or through direct comparison with natural quantum systems [83] [82]. This verifiability is a necessary condition for real-world utility, as it ensures computational results can be trusted and validated, much like established scientific instruments [84].
Central to this advancement is the development of the Quantum Echoes algorithm by Google Quantum AI, which leverages Out-of-Time-Order Correlators (OTOCs) to probe quantum systems with unprecedented sensitivity [83] [85]. This approach has demonstrated a 13,000x speedup over classical supercomputers while producing physically meaningful observables relevant to molecular simulation and materials science [86].
Theoretical Foundations: OTOCs and Quantum Chaos
Out-of-Time-Order Correlators (OTOCs)
Out-of-Time-Order Correlators are a specialized class of quantum observables that describe how quantum dynamics become chaotic. Unlike conventional measurements that follow a linear time sequence, OTOCs correlate operators at multiple time points in a non-chronological order, making them exceptionally sensitive to the underlying dynamics of quantum systems [83] [85].
The fundamental OTOC circuit implements a time-reversal protocol: a forward evolution (U), followed by a perturbation (B), then backward evolution (U†), and finally a measurement probe (M) [83]. Mathematically, for a k-th order OTOC, this process can be represented as:
[ \mathcal{C}^{(2k)} = \langle Uk^\dagger(t) M Uk(t) M \rangle = \langle (B(t)M)^{2k} \rangle ]
where ( B(t) = U^\dagger(t) B U(t) ) is the time-evolved perturbation operator [85].
Table 1: OTOC Orders and Their Characteristics
| OTOC Order | Circuit Structure | Key Feature | Classical Complexity |
|---|---|---|---|
| First-order (OTOC(1)) | Single round-trip evolution | Basic butterfly effect | More tractable |
| Second-order (OTOC(2)) | Double round-trip evolution | Constructive interference | Highly intractable |
| Higher-order (OTOC(k)) | k round-trip evolutions | Enhanced sensitivity | Extremely intractable |
Quantum Chaos and Information Scrambling
In chaotic quantum systems, quantum information becomes scrambled—dispersed throughout the system through entanglement, making most conventional observables insensitive to microscopic details over time [85] [86]. OTOCs uniquely maintain sensitivity in these regimes by leveraging quantum echoes to refocus scrambled information [85].
The interferometric nature of OTOCs leads to two crucial consequences for quantum advantage:
- Signal Amplification: The forward and backward evolutions partially reverse the effects of chaos, with OTOC signal magnitude decaying as a slow power law over time, compared to the exponential decay of conventional measurements [83].
- Classical Intractability: OTOCs manifest many-body interference effects that require tracking complex probability amplitudes across exponentially large spaces, presenting fundamental obstacles for classical simulation algorithms like quantum Monte Carlo [83].
The Quantum Echoes Algorithm: Implementation and Methodology
Core Algorithmic Framework
The Quantum Echoes algorithm implements OTOC measurement on quantum hardware through a carefully structured protocol that functions as a form of quantum interferometry [83] [86]. The algorithm can be conceptualized as a highly sensitive "quantum sonar" that sends signals into a quantum system and measures the returning echoes to reveal internal structure [84].
Figure 1: Quantum Echoes Algorithm Workflow - The complete OTOC measurement protocol showing forward evolution, perturbation, backward evolution, and measurement phases.
Experimental Protocol on Superconducting Qubits
Google's implementation on the Willow quantum chip utilized 65 of its 103 available superconducting qubits in a lattice configuration [83] [85] [86]. The experimental methodology followed this precise protocol:
Circuit Instance Generation: For each experimental run, researchers fixed the locations of the measurement qubit (qₘ) and perturbation qubit (q_b), then generated specific circuit instances by varying random parameters in single-qubit gates that interleaved deterministic two-qubit gates [85].
Echo Sequence Execution: The quantum system undergoes:
- Forward evolution (U) using random quantum circuits composed of single-qubit and fixed two-qubit gates
- Butterfly perturbation (B) applied to a specific qubit
- Backward evolution (U†) precisely reversing the forward evolution
- Probe operation (M) and final measurement [85]
Statistical Averaging: Each circuit instance was measured repeatedly until statistical noise fell below 10% of the average value, with the protocol repeated across 50-250 different circuit instances to establish statistical significance [85].
Error Mitigation: All experimental OTOC values were normalized using global rescaling factors derived from comprehensive error-mitigation strategies to account for hardware imperfections [85].
Table 2: Quantum Hardware Specifications for OTOC Experiments
| Parameter | Specification | Significance |
|---|---|---|
| Processor | Google Willow Chip | Superconducting quantum processor |
| Qubit Count | 65 qubits (of 103) | Beyond classical simulation capacity |
| Gate Error | Median 0.15% for two-qubit gates | Enables deep circuit execution |
| Circuit Depth | Up to 40 cycles | Sufficient for chaotic dynamics |
| Key Innovation | High-speed operations with low error rates | Enables precise time-reversal |
The Scientist's Toolkit: Essential Research Components
Table 3: Key Experimental Components for OTOC Research
| Component | Function | Implementation in Quantum Echoes |
|---|---|---|
| Time-Reversal Circuits | Reverse quantum evolution | Precisely implemented backward unitary U† |
| Butterfly Perturbation (B) | Initiates quantum butterfly effect | Single-qubit operation between evolutions |
| Probe Operator (M) | Extracts system response | Single-qubit measurement operation |
| Random Circuit Ensemble | Ensures quantum ergodicity | Parameterized single-qubit gates |
| Error Mitigation Protocols | Compensates for hardware noise | Global rescaling and statistical methods |
Verifiable Quantum Advantage: Experimental Results
Beyond-Classical Performance Metrics
The Quantum Echoes experiment demonstrated unambiguous quantum advantage through multiple quantitative metrics:
- Computational Speed: The 65-qubit OTOC(2) measurement required approximately 2.1 hours on the Willow processor, compared to an estimated 3.2 years on the Frontier supercomputer—a 13,000x speedup [83] [86].
- Classical Intractability: Researchers conducted extensive "red teaming" by implementing nine different classical simulation algorithms, including tensor-network contraction and quantum Monte Carlo, confirming the impossibility of predicting second-order OTOC data with classical methods [83].
- Signal Persistence: The standard deviation of OTOC(2) values decayed algebraically over time, remaining above 0.01 beyond 20 circuit cycles, while conventional time-ordered correlators decayed exponentially and became immeasurably small after just 9 cycles [85].
Verification Mechanisms
A crucial innovation of this approach is its built-in verification, which addresses a fundamental limitation of earlier quantum advantage demonstrations [82]:
- Cross-Quantum Verification: Results can be replicated on different quantum computers of similar capability, producing identical expectation values [83] [87].
- Nature-Based Validation: Predictions can be directly compared against experimental data from natural quantum systems, such as molecular NMR spectroscopy [83] [82].
Application to Molecular Hamiltonian Ground State Problems
Hamiltonian Learning Framework
The OTOC framework provides a powerful approach for Hamiltonian learning—extracting unknown parameters that govern quantum systems [83] [86]. The methodology involves:
- Quantum Simulation: Using the quantum computer to simulate OTOC signals from a physical system (e.g., molecules) with unknown parameters.
- Experimental Comparison: Comparing quantum computer OTOC signals against real-world experimental data.
- Parameter Optimization: Identifying system parameters that maximize agreement between simulation and experiment [83].
This approach is particularly valuable for molecular systems where precise Hamiltonian parameters are unknown but experimental NMR data is available.
Nuclear Magnetic Resonance (NMR) Enhancement
In proof-of-concept experiments with UC Berkeley, researchers applied Quantum Echoes to enhance nuclear magnetic resonance (NMR) spectroscopy, a fundamental tool for molecular structure determination [87]. Traditional NMR sensitivity drops sharply with distance between atomic nuclei, limiting measurable interactions. The OTOC protocol effectively creates a "longer molecular ruler" that can detect interactions between spins separated further apart [86].
Figure 2: Hamiltonian Learning via OTOC Matching - Experimental and computational pathways for determining molecular Hamiltonian parameters through OTOC comparison.
In demonstrated applications, researchers measured OTOCs on organic molecules dissolved in liquid crystal, then simulated these experiments on the Willow chip, resulting in improved molecular structure models [83]. While this initial demonstration remains within classical simulation capabilities, it establishes a pathway toward quantum advantage in molecular simulation.
Integration with Quantum Chemistry Workflows
The OTOC approach complements existing quantum computational chemistry methods like the Variational Quantum Eigensolver (VQE) by providing an alternative methodology for Hamiltonian parameter estimation [8] [9]. Recent research has also explored hybrid quantum-classical frameworks that embed quantum processors within larger-scale classical simulations, such as QM/MM (Quantum Mechanics/Molecular Mechanics) methods [28]. These integrative approaches position OTOC-based methods as valuable components in a broader toolkit for computational chemistry on emerging quantum hardware.
The demonstration of verifiable quantum advantage through OTOCs and the Quantum Echoes algorithm represents a watershed moment for quantum computation. By moving beyond abstract computational tasks to physically meaningful, verifiable observables, this approach establishes a viable path toward practical quantum applications in molecular science.
For researchers focused on molecular Hamiltonian ground state searches, OTOCs offer:
- A verification framework for quantum computations through cross-device reproducibility and nature-based validation
- Enhanced sensitivity to molecular parameters through interferometric amplification
- Hamiltonian learning capabilities that complement existing ground state preparation techniques
- A scalable path toward quantum utility in molecular simulation
As quantum hardware continues to improve, with efforts focused on achieving error-corrected logical qubits [87], the integration of OTOC-based methods with established quantum chemistry algorithms promises to unlock new capabilities for understanding and designing molecular systems. The era of verifiable quantum advantage marks not just a technical milestone but the beginning of quantum computation as a trustworthy scientific tool for molecular exploration.
The calculation of molecular ground-state energies represents a fundamental challenge in quantum chemistry, with profound implications for drug discovery and materials science. For decades, classical computational methods have faced exponential scaling walls when simulating quantum mechanical systems. The emergence of quantum computing offers a paradigm shift, promising to simulate nature using its own quantum-mechanical rules. This technical analysis provides a comprehensive comparison of computational scaling and resource requirements for molecular Hamiltonian ground-state searches, examining both established classical heuristics and emerging quantum algorithms within the context of current hardware capabilities and theoretical developments.
Fundamental Computational Principles
Classical Computing Paradigm
Classical computers process information using bits that exist in definite states of 0 or 1, executing operations sequentially through logical gates based on Boolean algebra. In computational chemistry, this architecture forces the approximation of quantum states using classical probability distributions, requiring exponential memory and processing resources to represent entangled quantum systems accurately. The fundamental limitation stems from the fact that representing an n-qubit quantum system requires storing ~2n complex numbers classically, creating an insurmountable scaling problem for simulating large molecular systems [88].
Quantum Computing Paradigm
Quantum computers leverage three fundamental phenomena—superposition, entanglement, and interference—to process information in ways fundamentally different from classical systems. Qubits can exist in coherent superpositions of |0⟩ and |1⟩ states, enabling parallel evaluation of multiple possibilities. Entanglement creates non-classical correlations between qubits, allowing for compact representation of molecular wavefunctions. For ground-state problems, this means quantum computers can naturally represent molecular Hamiltonians with resources that scale polynomially with system size, potentially bypassing the exponential bottlenecks of classical approaches [89].
Table 1: Fundamental Computational Properties
| Property | Classical Computing | Quantum Computing | ||
|---|---|---|---|---|
| Basic Unit | Bit (0 or 1) | Qubit (α | 0⟩ + β | 1⟩) |
| Information Representation | Definite states | Superposition states | ||
| Correlation Mechanism | Classical probability | Quantum entanglement | ||
| Algorithm Execution | Sequential operations | Parallel amplitude manipulation | ||
| Natural System Modeling | Classical physics | Quantum mechanics |
Classical Computational Approaches
Established Classical Heuristics
Classical computational chemistry employs sophisticated heuristics to approximate solutions to the electronic Schrödinger equation while avoiding full configuration interaction's exponential cost:
Density Matrix Embedding Theory (DMET): This fragmentation approach divides molecular systems into smaller, manageable subsystems embedded within an approximate electronic environment, enabling more scalable simulations while maintaining accuracy for weakly correlated regions [40].
Coupled Cluster Methods: The CCSD(T) approach—considered the "gold standard" in quantum chemistry—provides high accuracy for molecular energy predictions but scales as O(N7) with system size, rapidly becoming prohibitive for larger molecules [2].
Variational Monte Carlo: These methods use stochastic sampling to estimate quantum expectation values, providing favorable scaling compared to exact methods but introducing statistical uncertainties that must be carefully managed [2].
Classical Resource Requirements and Scaling
The computational resources required for classical ground-state determination depend critically on the desired accuracy and molecular size:
Table 2: Classical Method Scaling and Resource Requirements
| Method | Time Scaling | Memory Scaling | Accuracy | Application Domain |
|---|---|---|---|---|
| Exact Diagonalization | O(2^N) | O(2^N) | Exact | Small molecules (<~20 orbitals) |
| CCSD(T) | O(N^7) | O(N^4) | ~1 kcal/mol | Medium molecules |
| Density Functional Theory | O(N^3) | O(N^2) | ~3-5 kcal/mol | Large systems |
| DMET | O(N^3) (fragment-dependent) | O(N^2) | ~1-2 kcal/mol (fragment-dependent) | Large, weakly correlated systems |
For hardware configuration, classical molecular dynamics and electronic structure calculations typically utilize high-performance computing systems with 32-64 core CPUs (prioritizing clock speed over core count), coupled with multiple high-end GPUs (such as NVIDIA RTX 4090 or professional Ada series GPUs) for accelerated computation. Memory requirements typically range from 64GB to 256GB RAM, with sufficient NVMe storage for handling large wavefunction files [90].
Quantum Computational Approaches
Quantum Algorithms for Ground-State Energy Estimation
Quantum approaches to the molecular ground-state problem leverage problem-specific ansatzes and hybrid quantum-classical optimization strategies:
Variational Quantum Eigensolver (VQE): This hybrid algorithm prepares a parameterized ansatz state |ψ(θ)⟩ on a quantum processor, measures the expectation value ⟨ψ(θ)|H|ψ(θ)⟩, and uses classical optimization to minimize the energy. The ADAPT-VQE variant dynamically constructs ansatzes by iteratively adding operators from a predefined pool based on gradient contributions [4].
K-ADAPT-VQE Enhancement: This improvement reduces quantum resource requirements by adding operators in chunks of size K during each iteration, significantly reducing the total number of optimization cycles and quantum measurements needed to achieve chemical accuracy [4].
Quantum Phase Estimation (QPE): As a fault-tolerant algorithm, QPE projects an initial state onto Hamiltonian eigenstates via controlled time evolution, providing exponential speedup potential but requiring deep circuits and error correction currently unavailable on NISQ devices [2].
Quantum Resource Requirements and Scaling
The quantum resources for ground-state estimation are characterized by qubit counts, circuit depths, and measurement overheads:
Table 3: Quantum Algorithm Resource Requirements
| Algorithm | Qubit Count | Circuit Depth | Error Mitigation | Measurement Overhead | ||
|---|---|---|---|---|---|---|
| VQE | O(N) | Shallow | Error mitigation | Polynomial in parameters | ||
| ADAPT-VQE | O(N) | Adaptive | Error mitigation | Reduced iterations | ||
| K-ADAPT-VQE | O(N) | Adaptive | Error mitigation | Fewer optimization cycles | ||
| QPE | O(N) + ancillas | Deep (O(1/ϵ)) | Error correction | Poly(1/S) with S = | ⟨Φ∣Ψ₀⟩ |
For practical implementations, the DMET-SQD hybrid approach has demonstrated the capability to simulate molecular fragments using 27-32 qubits on IBM's Eagle processors, achieving chemical accuracy (within 1 kcal/mol) for systems like hydrogen rings and cyclohexane conformers through advanced error mitigation techniques including gate twirling and dynamical decoupling [40].
Comparative Scaling Analysis
Theoretical Scaling Comparison
The fundamental distinction between classical and quantum approaches emerges in their scaling behavior with system size. While exact classical methods exhibit exponential scaling O(2^N) for N orbitals, quantum algorithms promise polynomial scaling O(poly(N)) for the same problems. However, this theoretical advantage must be evaluated in the context of constant factors, state preparation costs, and precision requirements [2].
For quantum phase estimation, the total cost to obtain energy E to precision ϵ is poly(1/S)[poly(L)poly(1/ϵ) + C], where S represents the overlap |⟨Φ|Ψ₀⟩| between initial and target states, and C reflects state preparation complexity. This reveals that potential exponential advantage depends critically on maintaining polynomial scaling in the state preparation term, which cannot benefit from the same problem structure that enables efficient classical heuristic solutions [2].
Empirical Performance Evidence
Recent studies question whether exponential quantum advantage exists for generic chemical problems. Numerical investigations of iron-sulfur clusters (including nitrogenase FeMo-cofactor) examined the behavior of quantum state preparation strategies, finding that features enabling efficient heuristic quantum state preparation might similarly benefit classical heuristics [2].
The "orthogonality catastrophe" presents a particular challenge for quantum approaches: for O(L) non-interacting subsystems, the global overlap S between simple product states and the true ground-state decreases as s^O(L), potentially requiring exponential repetition overhead in QPE unless increasingly sophisticated initial states are prepared [2].
Implementation Pathways
Hardware Landscape
Classical Hardware: Specialized classical workstations for molecular simulation prioritize high-clock-speed CPUs (AMD Threadripper PRO or Intel Xeon W-3400 series) with 32-64 cores, coupled with multiple high-end GPUs (NVIDIA RTX 4090/6000 Ada) for accelerated computation. Memory configurations of 64-256GB RAM accommodate large wavefunction storage, with fast NVMe storage ensuring efficient data handling [90].
Quantum Hardware: Current quantum processors like IBM's 127-qubit Eagle and 120-qubit Nighthawk chips demonstrate improving metrics, with Heron processors achieving two-qubit gate errors below 1e-3 and execution speeds of 330,000 circuit layer operations per second (CLOPS). Error suppression techniques including probabilistic error cancellation (PEC) with 100x reduced sampling overhead and dynamical decoupling provide crucial mitigation of NISQ-era limitations [91].
Experimental Workflows
Diagram 1: VQE Workflow
Diagram 2: Resource Scaling
Research Reagent Solutions
Table 4: Essential Research Tools and Platforms
| Tool/Platform | Type | Primary Function | Application Context |
|---|---|---|---|
| IBM Quantum Platform | Hardware/Software | Quantum circuit execution | NISQ-era algorithm testing |
| Qiskit SDK | Software Framework | Quantum circuit design | Quantum algorithm development |
| PySCF | Classical Software | Electronic structure | Hamiltonian preparation |
| OpenFermion | Software Library | Fermion-to-qubit mapping | Quantum simulation interface |
| DMET-SQD Hybrid | Algorithmic Framework | Fragment embedding | Large molecule simulation |
| K-ADAPT-VQE | Algorithm | Adaptive ansatz construction | Resource-efficient VQE |
Future Outlook and Research Directions
The quantum-classical hybrid approach represents the most promising near-term path for molecular ground-state calculations. IBM's roadmap targets 5,000-gate circuits by end-of-2025 with their Nighthawk processor, while error correction milestones include RelayBP decoding achieving 480ns latency on AMD FPGAs [91]. As hardware improves, problems currently at the edge of classical tractability—such as transition metal cluster catalysis or excited-state dynamics—will become increasingly accessible to quantum simulation.
The research community is actively developing application libraries targeting Hamiltonian simulation, optimization, machine learning, and differential equations, with expected maturation by 2027. These libraries will leverage quantum-centric supercomputing architectures that integrate quantum and classical resources, enabling researchers to exploit each platform's respective strengths [91].
The integration of advanced computational methods with traditional experimental science is fundamentally reshaping drug discovery. Artificial intelligence (AI) and quantum computing have progressed from theoretical curiosities to powerful tools that can compress discovery timelines and address previously "undruggable" targets [92] [93]. This technical guide examines the integrated workflow from in-silico prediction to wet-lab confirmation, framed within the context of quantum computing for molecular Hamiltonian ground state search—a critical capability for accurately predicting molecular behavior in drug discovery.
The traditional drug discovery process typically requires approximately 5 years for discovery and preclinical work alone, with costs reaching $1-3 billion per approved drug [92] [94]. AI-driven platforms have demonstrated the potential to reduce early-stage discovery from years to months in some cases, with Exscientia reporting 70% faster design cycles requiring 10-fold fewer synthesized compounds than industry norms [92]. Meanwhile, quantum computing presents a multibillion-dollar opportunity to revolutionize discovery by enabling accurate molecular simulations that are intractable for classical computers [95].
This paradigm shift replaces labor-intensive, human-driven workflows with AI-powered discovery engines capable of compressing timelines, expanding chemical and biological search spaces, and redefining the speed and scale of modern pharmacology [92]. However, the ultimate validation of any computational prediction remains rigorous experimental confirmation, creating the essential bridge between digital innovation and therapeutic reality.
Quantum Computing Advances for Molecular Systems
Quantum Approaches to Molecular Hamiltonian Ground State Problems
The accurate determination of molecular ground state energies is fundamental to drug discovery, as these energies dictate molecular stability, reactivity, and binding interactions. Quantum computational chemistry leverages qubits to represent molecular wavefunctions, potentially simulating quantum systems more efficiently than classical methods [75]. The Variational Quantum Eigensolver (VQE) algorithm has emerged as the predominant framework for these calculations, variationally learning the quantum state of molecular systems [75].
Recent research has introduced innovative hybrid frameworks that combine quantum circuits with classical computational approaches to overcome significant limitations in accuracy due to hardware noise and algorithmic constraints. The pUNN (paired unitary coupled-cluster with neural networks) method exemplifies this approach, employing a linear-depth paired Unitary Coupled-Cluster with double excitations (pUCCD) circuit to learn molecular wavefunction in the seniority-zero subspace, while a neural network accounts for contributions from unpaired configurations [75]. This hybrid quantum-neural wavefunction approach maintains low qubit count (N qubits) and shallow circuit depth while achieving accuracy comparable to advanced quantum and classical computational chemistry methods like UCCSD and CCSD(T) [75].
Enhanced Precision on Near-Term Quantum Hardware
Achieving chemical precision (1.6 × 10−3 Hartree) on current quantum devices requires overcoming significant challenges including readout errors, limited sampling, and circuit depth constraints. Recent research has implemented practical techniques to address these limitations:
- Locally biased random measurements reduce shot overhead (the number of quantum computer measurements) by prioritizing measurement settings with greater impact on energy estimation [78].
- Repeated settings with parallel quantum detector tomography (QDT) mitigate readout errors and reduce circuit overhead (the number of different gate implementations) [78].
- Blended scheduling accounts for dynamic, time-dependent noise by interleaving circuits for different Hamiltonians with calibration circuits [78].
When applied to molecular energy estimation of the BODIPY molecule on a Hartree-Fock state, these techniques collectively reduced measurement errors by an order of magnitude—from 1-5% to 0.16% on an IBM Eagle r3 quantum processor [78]. This enhanced precision is particularly valuable for drug discovery applications where accurate energy calculations are essential for predicting molecular interactions.
Quantum Machine Learning for Drug-Target Interactions
Beyond quantum chemistry simulations, quantum computing enhances machine learning models for predicting drug-target interactions (DTI). In a landmark study, researchers at St. Jude and the University of Toronto developed a hybrid pipeline that alternated between training classical and quantum machine learning models to generate novel molecules predicted to bind to the challenging KRAS protein, a frequent cancer driver historically considered "undruggable" [93].
This approach successfully identified two novel KRAS-binding molecules that were subsequently validated through experimental testing, representing the first experimental validation of quantum computing in drug discovery [93]. The quantum model outperformed similar purely classical machine learning models, demonstrating quantum computing's potential to identify therapeutic compounds for difficult targets.
Table 1: Quantum Computing Applications in Drug Discovery
| Application Area | Technical Approach | Key Achievement | Experimental Validation |
|---|---|---|---|
| Molecular Energy Estimation | Hybrid quantum-neural wavefunction (pUNN) | Near-chemical accuracy for molecular systems | Cyclobutadiene isomerization reaction simulated on superconducting quantum processor [75] |
| Precision Enhancement | Quantum detector tomography + blended scheduling | Error reduction to 0.16% on IBM Eagle r3 | BODIPY molecule energy estimation [78] |
| Drug-Target Interaction | Quantum machine learning model fusion | Novel KRAS-binding molecules identified | Two molecules experimentally validated for KRAS binding [93] |
| Protein-Ligand Simulation | Quantum-accelerated computational chemistry | Cytochrome P450 simulation with precision beyond classical methods | Collaboration between Google and Boehringer Ingelheim [15] |
Integrated Experimental Validation Workflow
The transition from in-silico prediction to wet-lab confirmation requires a systematic, integrated workflow. The following diagram illustrates this end-to-end process, highlighting the critical feedback loops between computational and experimental phases:
Figure 1: Integrated Quantum-Classical Drug Discovery Workflow with Experimental Validation Feedback Loops
Computational Screening and Prioritization Protocol
The initial computational phase employs multi-tier screening to prioritize candidates for experimental testing:
Step 1: Target Identification and Compound Library Preparation
- Identify therapeutic targets through analysis of genomic, proteomic, and clinical data using AI systems [94]
- Prepare virtual compound libraries (existing libraries contain >11 billion compounds) [94]
- Apply pharmacophore modeling and QSAR to define essential interaction features [96]
Step 2: Quantum-Enhanced Screening
- Employ quantum computing for first-principles calculations of molecular interactions [95]
- Implement molecular docking simulations to predict binding affinities [94]
- Use hybrid quantum-classical algorithms like VQE or quantum machine learning for initial screening [93] [75]
Step 3: Classical AI Refinement
- Apply evidential deep learning (EviDTI) for uncertainty quantification to prioritize candidates with reliable predictions [97]
- Integrate multi-dimensional data including drug 2D topological graphs, 3D spatial structures, and target sequence features [97]
- Use network-based protocols to map biologically relevant chemical space and identify structural analogs [98]
Experimental Validation Methodologies
Primary Binding Assays
- Disk diffusion assays: Qualitative assessment of antibacterial activity (e.g., for anti-Staphylococcus aureus compounds) [98]
- Resazurin-based microdilution assays: Quantitative measurement of metabolic inhibition through fluorescence/absorbance [98]
- Surface plasmon resonance (SPR): Label-free quantification of binding kinetics and affinity
Functional and Selectivity Profiling
- Cell viability assays (MTT/XTT): Cytotoxicity evaluation against target and normal cell lines
- High-content phenotypic screening: Ex vivo testing on patient-derived samples (e.g., Exscientia's patient-first approach) [92]
- Kinase profiling panels: Selectivity assessment against related off-targets
ADME-Tox Prediction and Validation
- In vitro metabolic stability assays: Microsomal/hepatocyte incubation with LC-MS quantification
- CYP450 inhibition screening: Potential drug-drug interaction assessment
- Plasma protein binding: Equilibrium dialysis or ultrafiltration methods
- In silico toxicity prediction: Quantum calculations of structural transformations influencing toxicity [94]
Table 2: Experimental Validation Techniques for Computational Predictions
| Validation Type | Experimental Method | Key Output Parameters | Typical Timeline |
|---|---|---|---|
| Binding Affinity | Resazurin microdilution | MIC values, metabolic inhibition fold-reduction | 1-2 weeks |
| Target Engagement | Cellular thermal shift assay (CETSA) | Target thermal stability, binding confirmation | 2-3 weeks |
| Functional Activity | Pathway reporter assays | IC50/EC50, signaling modulation | 2-4 weeks |
| Selectivity | Kinase profiling panels | Selectivity scores, off-target identification | 3-4 weeks |
| ADME | Metabolic stability assay | Intrinsic clearance, half-life | 2-3 weeks |
| Cytotoxicity | MTT/XTT cell viability | CC50, therapeutic index | 1-2 weeks |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful experimental validation requires carefully selected reagents and materials. The following table details essential components for establishing these assays:
Table 3: Essential Research Reagent Solutions for Experimental Validation
| Reagent/Material | Function and Application | Example Specifications |
|---|---|---|
| Resazurin sodium salt | Metabolic activity indicator in microdilution assays; measures bacterial viability through fluorescence conversion | 0.02% solution in PBS; excitation 530-560nm, emission 590nm [98] |
| Cacalol derivatives | Natural product-derived compounds for anti-Staphylococcus aureus activity validation | 20mM stock in DMSO; 7-10mm inhibition zones in disk diffusion [98] |
| KRAS protein mutants | Target protein for validating quantum machine learning predictions in cancer drug discovery | G12C, G12D, G12V mutants; full-length or binding domain [93] |
| Patient-derived tumor samples | Ex vivo phenotypic screening for translational relevance assessment | Fresh or cryopreserved samples; 3D organoid cultures preferred [92] |
| Cytochrome P450 isoforms | Metabolic stability and drug interaction profiling | Human liver microsomes or recombinant CYP enzymes [15] |
| AlphaFold-predicted structures | Structural templates for molecular docking and target analysis | Predicted local distance difference test (pLDDT) >70 for reliable regions [96] |
| Quantum computing cloud access | Quantum simulation of molecular Hamiltonians and machine learning | IBM Quantum, Amazon Braket, or Rigetti Quantum Cloud Services [92] [15] |
Technical Protocols for Key Experiments
Protocol 1: Resazurin-Based Microdilution Assay for Antibacterial Validation
This protocol provides quantitative confirmation of antibacterial activity for computationally-predicted compounds [98].
Materials Preparation
- Prepare resazurin solution (0.02% in phosphate-buffered saline)
- Dilute test compounds to 20mM in DMSO (or appropriate solvent)
- Culture Staphylococcus aureus strains to mid-log phase (OD600 ≈ 0.5)
Procedure
- Prepare serial dilutions of test compounds in 96-well plates (final volume 100μL)
- Add bacterial inoculum (5×10^5 CFU/well) to all test wells
- Include growth control (bacteria + solvent), sterility control (media only), and reference antibiotic controls
- Incubate plates at 37°C for 18-24 hours
- Add 20μL resazurin solution to each well and incubate 2-4 hours
- Measure fluorescence (excitation 530-560nm, emission 590nm) or visual color change
Data Analysis
- Calculate percentage reduction in metabolism compared to growth control
- Determine minimum inhibitory concentration (MIC) as lowest concentration showing ≥80% inhibition
- Validate with disk diffusion assays (7-10mm inhibition zones indicate significant activity) [98]
Protocol 2: Quantum-Classical Hybrid Model Validation for KRAS Targeting
This protocol outlines the experimental validation of quantum machine learning predictions for challenging drug targets [93].
Computational Phase
- Train classical machine learning model on known KRAS binders and ultra-large virtual screening data (>100,000 theoretical binders)
- Develop quantum machine learning filter/reward function to evaluate generated molecule quality
- Alternately train classical and quantum models to optimize in concert
- Generate novel ligand structures predicted to bind KRAS mutants
Experimental Validation Phase
- Express and purify KRAS protein (wild-type and common mutants)
- Synthesize or source top-predicted compounds (typically 10-50 candidates)
- Perform binding affinity measurements (SPR or thermal shift assays)
- Conduct cellular efficacy studies in KRAS-mutant cancer lines
- Evaluate selectivity against related GTPases (HRAS, NRAS)
Success Criteria
- Confirmed binding to KRAS with Kd < 10μM
- Cellular activity in KRAS-dependent models with IC50 < 10μM
- Minimum 10-fold selectivity over related targets
The following diagram illustrates the technical workflow for this hybrid validation approach:
Figure 2: Quantum-Classical Hybrid Workflow for KRAS Ligand Discovery and Validation
The integration of quantum computing, AI, and experimental validation represents a transformative approach to drug discovery. By leveraging quantum computers for molecular Hamiltonian ground state searches and molecular simulations, researchers can explore chemical space with unprecedented accuracy and efficiency [95] [75]. The emerging paradigm of hybrid quantum-classical algorithms, combined with rigorous experimental validation, creates a powerful feedback loop that accelerates the identification and optimization of therapeutic compounds.
As quantum hardware continues to advance with improvements in error correction and qubit coherence times [15], and as AI methodologies incorporate better uncertainty quantification through approaches like evidential deep learning [97], the reliability of in-silico predictions will further improve. This progress will enable researchers to prioritize the most promising candidates for experimental validation, ultimately reducing the time and cost of bringing new therapeutics to patients while addressing previously intractable targets [93] [94].
The future of drug discovery lies in the seamless integration of computational prediction and experimental confirmation—where quantum simulations inform wet-lab experiments, and experimental results refine computational models. This virtuous cycle promises to unlock new therapeutic possibilities and reshape the landscape of pharmaceutical development in the coming years.
Conclusion
The search for molecular Hamiltonian ground states is rapidly transitioning from a theoretical pursuit to a practical application of quantum computing, marked by verified speedups and promising hybrid methods. As outlined, foundational principles are being translated into robust methodologies capable of running on today's hardware, while sophisticated error mitigation and algorithmic co-design are steadily overcoming the limitations of the NISQ era. The recent demonstrations of verifiable quantum advantage, achieving results thousands of times faster than classical supercomputers, signal a pivotal shift. For biomedical research, this progress heralds a future where quantum computers will dramatically accelerate drug discovery, enable the precise simulation of complex biological systems like previously 'undruggable' targets, and ultimately pave the way for a new era of predictive, in-silico-driven therapeutic development. The continued convergence of improved hardware, advanced algorithms, and domain-specific applications promises to unlock unprecedented capabilities in understanding and manipulating matter at the quantum level.
References
- 1. Molecular Hamiltonian [https://en.wikipedia.org/wiki/Molecular_Hamiltonian]
- 2. Evaluating the evidence for exponential quantum ... [https://www.nature.com/articles/s41467-023-37587-6]
- 3. Reduction of the molecular hamiltonian matrix using ... [https://www.nature.com/articles/s41598-021-83561-x]
- 4. K-ADAPT-VQE: Optimizing Molecular Ground State ... [https://arxiv.org/html/2506.09658v1]
- 5. [2504.07568] Ground State Energy of He molecule Using a ... [https://arxiv.org/abs/2504.07568]
- 6. Quantum Chemistry — PennyLane [https://docs.pennylane.ai/en/stable/introduction/chemistry.html]
- 7. [2504.18156] Determining Molecular Ground State with ... [https://arxiv.org/abs/2504.18156]
- 8. Quantum computing for molecular vibrational energies [https://www.sciencedirect.com/science/article/pii/S2950257825000095]
- 9. Dissipative ground state preparation in ab initio electronic ... [https://www.nature.com/articles/s41534-025-01124-8]
- 10. Stabilizer ground states for simulating quantum many-body ... [https://quantum-journal.org/papers/q-2025-06-24-1782/]
- 11. electronic structure - How to overcome the exponential ... wall [https://mattermodeling.stackexchange.com/questions/64/how-to-overcome-the-exponential-wall-encountered-in-full-configurational-interac]
- 12. Quantum Computing Beyond Ground State Electronic ... [https://arxiv.org/html/2509.19709v1]
- 13. Will quantum computing be chemistry's next AI? - C&EN [https://cen.acs.org/business/quantum-computing-chemistrys-next-AI/103/web/2025/11]
- 14. Beyond the Hype: Quantum Computers Start Solving Real ... [https://viterbischool.usc.edu/news/2025/11/beyond-the-hype-quantum-computers-start-solving-real-problems/]
- 15. Quantum Computing Industry Trends 2025: A Year of ... [https://www.spinquanta.com/news-detail/quantum-computing-industry-trends-2025-breakthrough-milestones-commercial-transition]
- 16. Adiabatic state preparation from general initial states [https://arxiv.org/abs/2511.05689]
- 17. Variational quantum algorithm for molecular geometry ... [https://arxiv.org/abs/2106.13840]
- 18. Superposition and entanglement [https://www.quantum-inspire.com/kbase/superposition-and-entanglement/]
- 19. What Is Quantum Computing? Full Beginner Guide 2025 [https://www.spinquanta.com/news-detail/what-is-quantum-computing-and-how-does-it-work-expert-explained]
- 20. What Are Superposition & Entanglement in Quantum ... [https://www.dummies.com/article/technology/computers/what-are-superposition-entanglement-in-quantum-computing-300563/]
- 21. Building molecular Hamiltonians | PennyLane Demos [https://pennylane.ai/qml/demos/tutorial_quantum_chemistry]
- 22. What Is Quantum Computing? | IBM [https://www.ibm.com/think/topics/quantum-computing]
- 23. Consensus-based qubit configuration optimization for ... [https://www.nature.com/articles/s41534-025-01132-8]
- 24. Efficient Hamiltonian Simulation: A Utility Scale Perspective ... [https://arxiv.org/abs/2412.15804]
- 25. Quantum Computing Faces 3 Major Barriers Before Going ... [https://www.forbes.com/sites/bernardmarr/2025/07/23/quantum-computing-faces-3-major-barriers-before-going-mainstream/]
- 26. Quantum complexity theory [https://en.wikipedia.org/wiki/Quantum_complexity_theory]
- 27. BQP [https://en.wikipedia.org/wiki/BQP]
- 28. Extending quantum computing through subspace, embedding ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00225g]
- 29. Application to Ground-State Energy Estimation [https://quantum-journal.org/papers/q-2025-04-01-1682/]
- 30. Evidence for the utility of quantum computing before fault ... [https://www.nature.com/articles/s41586-023-06096-3]
- 31. 5-1. Variational Quantum Eigensolver (VQE) Algorithm [https://dojo.qulacs.org/en/latest/notebooks/5.1_variational_quantum_eigensolver.html]
- 32. Variational quantum eigensolver [https://en.wikipedia.org/wiki/Variational_quantum_eigensolver]
- 33. A brief overview of VQE | PennyLane Demos [https://pennylane.ai/qml/demos/tutorial_vqe]
- 34. Calculating the ground state energy of molecules [https://medium.com/@ketanmore1137/calculating-the-ground-state-energy-of-molecules-ba2a8be0a599]
- 35. a quantum Krylov algorithm with near-optimal sampling cost [https://arxiv.org/abs/2511.20998]
- 36. Measurement-efficient quantum Krylov subspace ... [https://quantum-journal.org/papers/q-2024-08-13-1438/]
- 37. Intro to Quantum Phase Estimation [https://pennylane.ai/qml/demos/tutorial_qpe]
- 38. What is Quantum Phase Estimation [https://www.quera.com/glossary/quantum-phase-estimation]
- 39. Filtered Quantum Phase Estimation [https://arxiv.org/html/2510.04294]
- 40. Study Suggests Today's Quantum Computers Could Aid ... [https://thequantuminsider.com/2025/07/15/study-suggests-todays-quantum-computers-could-aid-molecular-simulation/]
- 41. Towards quantum-centric simulations of extended molecules [https://arxiv.org/html/2411.09861v1]
- 42. Density matrix embedding theory [https://en.wikipedia.org/wiki/Density_matrix_embedding_theory]
- 43. Sample-based Krylov Quantum Diagonalization [https://arxiv.org/html/2501.09702v1]
- 44. Accurate quantum-centric simulations of intermolecular ... [https://www.nature.com/articles/s42005-025-02305-9]
- 45. [2502.02139] Fault-tolerant quantum algorithms for ... [https://arxiv.org/abs/2502.02139]
- 46. Variational preparation of normal matrix product states on ... [https://arxiv.org/html/2503.09683v3]
- 47. Introducing matrix product states for quantum practitioners [https://pennylane.ai/qml/demos/tutorial_mps]
- 48. Quantum state preparation of normal distributions using ... [https://www.nature.com/articles/s41534-024-00805-0]
- 49. Pauli Measurements in Quantum Computing [https://learn.microsoft.com/en-us/azure/quantum/concepts-pauli-measurements]
- 50. Adiabatic state preparation from general initial states [https://arxiv.org/html/2511.05689]
- 51. Quantum state preparation via piecewise QSVT [https://quantum-journal.org/papers/q-2025-07-03-1786/]
- 52. Quantum-machine-assisted Drug Discovery [https://arxiv.org/html/2408.13479v5]
- 53. A hybrid quantum computing pipeline for real world drug ... [https://www.nature.com/articles/s41598-024-67897-8]
- 54. Targeting KRAS mutations: orchestrating cancer evolution ... [https://www.nature.com/articles/s41392-025-02473-8]
- 55. Drugging the 'undruggable' KRAS: breakthroughs ... [https://www.cancerbiomed.org/content/early/2025/07/07/j.issn.2095-3941.2025.0122]
- 56. The State of KRAS Drug Development in Pancreatic Cancer [https://lustgarten.org/from-undruggable-to-unstoppable-the-state-of-kras-drug-development-in-pancreatic-cancer/]
- 57. Navigating the complexities of drug development for metallo-β ... [https://pubs.rsc.org/en/content/articlehtml/2025/md/d5md00035a]
- 58. About Error Mitigation [https://mitiq.readthedocs.io/en/stable/guide/error-mitigation.html]
- 59. Differentiating quantum error correction, suppression, and ... [https://q-ctrl.com/topics/differentiating-quantum-error-correction-suppression-and-mitigation]
- 60. Johns Hopkins Team Introduces New Method for Mapping ... [https://thequantuminsider.com/2025/11/21/johns-hopkins-maps-quantum-noise/]
- 61. Error mitigation with stabilized noise in superconducting ... [https://www.nature.com/articles/s41467-025-62820-9]
- 62. Differences in error suppression, mitigation, and correction [https://www.ibm.com/quantum/blog/quantum-error-suppression-mitigation-correction]
- 63. [2312.07497] Practical Benchmarking of Randomized ... [https://arxiv.org/abs/2312.07497]
- 64. QB Ground State Energy Estimation Benchmark [https://arxiv.org/abs/2508.10873]
- 65. Multireference Embedding and Fragmentation Methods for ... [https://arxiv.org/html/2505.13394v1]
- 66. Quantum Computing 2025: Beyond Hype to Real ... [https://medium.com/@daveshpandey/quantum-computing-2025-beyond-hype-to-real-breakthroughs-e45132b20e0c]
- 67. Alice & Bob Shortens Timeline to Quantum Computing ... [https://alice-bob.com/newsroom/alice-bob-quantum-computing-applications-health-agriculture/]
- 68. Co-Design [https://www.qscience.org/co-design/]
- 69. Co-Design Will Drive Industry-Specific Quantum Solutions [https://www.quera.com/blog-posts/prediction-co-design-will-drive-industry-specific-quantum-solutions-accelerating-development-of-application-ready-systems]
- 70. Co-Design Center for Quantum Advantage [https://ny-creates.org/quantum-technologies/co-design-center-for-quantum-advantage/]
- 71. Hardware-tailored diagonalization circuits | npj Quantum ... [https://www.nature.com/articles/s41534-024-00901-1]
- 72. Generative quantum combinatorial optimization by means of a ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00138b]
- 73. Post-processing [https://www.quantinuum.com/glossary-item/post-processing]
- 74. Classical post-processing approach for quantum amplitude ... [https://arxiv.org/abs/2502.05617]
- 75. Quantum machine learning of molecular energies with hybrid ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00222b]
- 76. BenchQC: A Benchmarking Toolkit for Quantum Computation [https://pmc.ncbi.nlm.nih.gov/articles/PMC12614196/]
- 77. BenchQC: A Benchmarking Toolkit for Quantum Computation [https://arxiv.org/html/2502.09595v1]
- 78. Practical techniques for high-precision measurements on ... [https://www.nature.com/articles/s41534-025-01066-1]
- 79. Extending quantum-mechanical benchmark accuracy to ... [https://www.nature.com/articles/s41467-025-63587-9]
- 80. Architectures, Initializations, and Optimizers for Silicon ... [https://arxiv.org/html/2510.23171v1]
- 81. Quantum Advantage in Computational Chemistry? † [https://arxiv.org/html/2508.20972v1]
- 82. The Grand Challenge of Quantum Applications [https://arxiv.org/html/2511.09124v1]
- 83. A verifiable quantum advantage [https://research.google/blog/a-verifiable-quantum-advantage/]
- 84. Google's New 'Quantum Echoes Algorithm' and My Last ... [https://edrm.net/2025/10/googles-new-quantum-echoes-algorithm-and-my-last-article-quantum-echo/]
- 85. Observation of constructive interference at the edge ... [https://www.nature.com/articles/s41586-025-09526-6]
- 86. Google Quantum AI Shows 13000× Speedup Over World's ... [https://thequantuminsider.com/2025/10/22/google-quantum-ai-shows-13000x-speedup-over-worlds-fastest-supercomputer-in-physics-simulation/]
- 87. Our Quantum Echoes algorithm is a big step toward real-world ... [https://chemistry.berkeley.edu/news/our-quantum-echoes-algorithm-big-step-toward-real-world-applications-quantum-computing]
- 88. Quantum Computing vs Classical Computing [https://www.spinquanta.com/news-detail/quantum-computing-vs-classical-computing-full-breakdown]
- 89. Quantum vs. Classical: Comparative Advantages in ... [https://www.spinquanta.com/news-detail/quantum-vs-classical-comparative-advantages-in-problem-solving]
- 90. Best CPU, GPU, RAM for Molecular Dynamics [https://www.sabrepc.com/blog/life-sciences/best-cpu-gpu-and-ram-for-md-workstation-server?srsltid=AfmBOoo766pyOpCPPO6dENnNakJJsW7RJUmakCnqRMTr7VUwsDg_BH5e]
- 91. Scaling for quantum advantage and beyond [https://www.ibm.com/quantum/blog/qdc-2025]
- 92. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 93. Quantum computing makes waves in drug discovery [https://www.stjude.org/research/progress/2025/quantum-computing-makes-waves-in-drug-discovery.html]
- 94. Harnessing AI and Quantum Computing for Revolutionizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12306909/]
- 95. Quantum computing in life sciences and drug discovery [https://www.mckinsey.com/industries/life-sciences/our-insights/the-quantum-revolution-in-pharma-faster-smarter-and-more-precise]
- 96. Computational-aided drug design strategies for drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12634332/]
- 97. Evidential deep learning-based drug-target interaction ... [https://www.nature.com/articles/s41467-025-62235-6]
- 98. Discovery of Anti-Staphylococcus aureus Cacalol Derivatives [https://www.frontiersin.org/journals/drug-discovery/articles/10.3389/fddsv.2025.1724392/full]