Variational Quantum Algorithms: A Practical Guide for Combinatorial Chemistry and Drug Discovery
This article provides a comprehensive introduction to Variational Quantum Algorithms (VQAs) and their transformative potential in combinatorial chemistry and drug discovery.
Variational Quantum Algorithms: A Practical Guide for Combinatorial Chemistry and Drug Discovery
Abstract
This article provides a comprehensive introduction to Variational Quantum Algorithms (VQAs) and their transformative potential in combinatorial chemistry and drug discovery. Aimed at researchers, scientists, and drug development professionals, it explores the foundational principles of VQAs like VQE and QAOA, detailing their application to critical tasks such as molecular energy calculations and optimizing complex chemical search spaces. The content addresses key practical challenges including the barren plateau problem and circuit depth limitations, while presenting validation strategies and performance comparisons with classical computational methods. By synthesizing current research and real-world case studies, this guide serves as a roadmap for integrating quantum computing into existing drug design workflows.
Quantum Foundations: Why VQAs are Suited for Chemical Problems
The advent of the Noisy Intermediate-Scale Quantum (NISQ) era marks a pivotal transition in computational science, particularly for fields requiring complex molecular simulation. Today's quantum processors, characterized by 50-1000 qubits with high error rates and limited coherence times, cannot yet support fault-tolerant quantum computation [1]. Gate fidelities currently hover around 99-99.5% for single-qubit operations and 95-99% for two-qubit gates, constraining circuit depth and complexity [1]. Within these hardware limitations, Variational Quantum Algorithms (VQAs) have emerged as the dominant algorithmic framework, offering a practical pathway to explore quantum advantage in combinatorial chemistry research by combining short-depth quantum circuits with classical optimization routines [2] [3].
This technical guide examines VQAs specifically through the lens of combinatorial chemistry applications, where simulating molecular systems and predicting chemical properties remains computationally challenging for classical computers. We provide researchers with both theoretical foundations and practical methodologies for implementing these hybrid quantum-classical algorithms, with particular emphasis on the Variational Quantum Eigensolver (VQE) and its variants for molecular energy calculations.
Theoretical Foundations of Variational Quantum Algorithms
Core Principles and Mathematical Framework
VQAs operate on a hybrid quantum-classical principle where parameterized quantum circuits (PQCs) serve as function evaluators while classical optimizers adjust parameters to minimize a cost function. The mathematical foundation rests on the Rayleigh-Ritz variational principle, which states that for any trial wavefunction (|\ψ(\theta)\rangle), the expectation value of the Hamiltonian (\hat{H}) provides an upper bound to the true ground state energy (E_0) [2]:
[ E_0 \leq \langle \psi(\theta) | \hat{H} | \psi(\theta) \rangle ]
The algorithm seeks to find parameters (\theta) that minimize this expectation value through iterative evaluation and adjustment [2]. This approach makes VQAs particularly suitable for NISQ devices because they accommodate shallow circuit depths and inherent resilience to certain noise types.
Key Algorithmic Components
Three essential components form the architecture of any VQA:
Parameterized Quantum Circuit (Ansatz): The ansatz (U(\theta)) comprises a sequence of parameterized unitary transformations applied to an initial quantum state. The choice of ansatz determines both expressibility and entanglement capacity, with hardware-efficient ansätze favoring NISQ implementation constraints while chemically-inspired ansätze (like UCCSD) provide more physically meaningful parameterizations [2].
Cost Function: The cost function (C(\theta)) represents the hypersurface to be minimized and is analogous to loss functions in classical machine learning. For quantum chemistry applications, this is typically the expectation value of the molecular Hamiltonian, often decomposed into measurable Pauli terms [2].
Classical Optimizer: Optimization routines range from gradient-based methods (using techniques like the parameter-shift rule for gradient estimation) to gradient-free approaches, with the choice heavily influencing convergence behavior and resilience to noise-induced barren plateaus [2].
Key Variational Quantum Algorithms for Chemistry
Variational Quantum Eigensolver (VQE)
The Variational Quantum Eigensolver (VQE) stands as the most successfully demonstrated VQA for quantum chemistry applications, specifically designed to find ground state energies of molecular systems [1]. The VQE optimization problem is formally expressed as:
[ E{VQE} = \min{\theta} \langle \phi | U^\dagger(\theta) \hat{H} U(\theta) | \phi \rangle ]
where (|\phi\rangle) represents the initial state, typically (|0\rangle^{\otimes n}) [2]. Experimental implementations have achieved chemical accuracy (<1.6 mHa) for small molecules including H₂, LiH, and BeH₂ using hardware-efficient ansätze on superconducting qubit platforms [2]. The UCCSD ansatz provides chemically meaningful parameters but produces deeper circuits more susceptible to noise [2].
Table 1: VQE Performance on Small Molecular Systems
| Molecule | Qubits Required | Ansatz Type | Achieved Accuracy | Hardware Platform |
|---|---|---|---|---|
| H₂ | 2 | Hardware-efficient | Chemical accuracy | Superconducting qubits [2] |
| LiH | 4-6 | UCCSD | Chemical accuracy | Superconducting qubits [2] |
| BeH₂ | 6-8 | UCCSD | Chemical accuracy | Superconducting qubits [2] |
| H₂O | 10-14 | UCCSD | Near chemical accuracy | Simulations [4] |
Quantum Approximate Optimization Algorithm (QAOA) and Chemistry Applications
While primarily designed for combinatorial optimization, the Quantum Approximate Optimization Algorithm (QAOA) can be adapted for quantum chemistry problems mapped to Ising models or QUBO formulations. QAOA constructs a quantum circuit with (p) alternating layers of problem and mixer Hamiltonians:
[ |\psi(\gamma, \beta)\rangle = \prod{j=1}^p e^{-i\betaj \hat{H}M} e^{-i\gammaj \hat{H}_C} |+\rangle^{\otimes n} ]
Classical optimization then adjusts the parameters ({\gammaj, \betaj}) to minimize the expectation value of the problem Hamiltonian (\hat{H}_C) [1]. For chemistry applications, this approach can be particularly useful for certain molecular similarity problems or combinatorial aspects of molecular design.
Advanced Variational Approaches
Recent research has expanded the VQA landscape with algorithms showing promise for chemical applications:
- Variational Quantum Imaginary Time Evolution (VarQITE): This approach simulates imaginary time evolution to prepare ground states and has demonstrated improved performance for certain problem classes compared to VQE [5].
- Parallelized VQAs: Framework for splitting quantum circuits to solve problems larger than available qubits, enabling larger chemical systems to be addressed through problem decomposition [6].
Experimental Protocols and Workflows
Standard VQE Protocol for Molecular Ground States
The following workflow represents the established methodology for calculating molecular ground state energies using VQE:
Diagram 1: VQE hybrid workflow (Adapted from [2])
Step-by-Step Protocol:
Hamiltonian Formulation: Transform the molecular electronic structure problem into a qubit Hamiltonian using Jordan-Wigner or Bravyi-Kitaev transformations, resulting in a weighted sum of Pauli terms: (\hat{H} = \sumi ci P_i) [4].
Ansatz Selection: Choose an appropriate parameterized quantum circuit architecture. Options include:
Parameter Initialization: Initialize parameters using random values, classical approximations, or heuristic methods to start the optimization process [2].
Quantum Processing: Execute the parameterized quantum circuit on quantum hardware or simulator to prepare the trial state (|\psi(\theta)\rangle = U(\theta)|0\rangle^{\otimes n}).
Expectation Value Measurement: Measure each Hamiltonian term (\langle P_i \rangle) through repeated circuit execution, requiring substantial sampling to achieve statistical precision [2].
Classical Optimization: Employ classical optimizers (e.g., gradient descent, SPSA, or NM) to adjust parameters (\theta) based on the computed energy (E(\theta) = \sumi ci \langle P_i \rangle).
Convergence Check: Iterate until energy changes fall below predetermined thresholds or maximum iterations reached, then validate against classical methods where possible.
Error Mitigation Techniques for Chemistry Calculations
NISQ-era computations require specialized error mitigation strategies to extract meaningful results:
- Zero-Noise Extrapolation (ZNE): Artificially amplifies circuit noise through stretching gate times or inserting identities, then extrapolates to the zero-noise limit [1].
- Symmetry Verification: Exploits conservation laws (particle number, spin symmetry) inherent in chemical systems to detect and discard erroneous measurements [1].
- Probabilistic Error Cancellation: Constructs ideal quantum operations as linear combinations of implementable noisy operations, though with significant sampling overhead [1].
Table 2: Error Mitigation Methods for Quantum Chemistry Calculations
| Technique | Mechanism | Sampling Overhead | Best-Suited Applications |
|---|---|---|---|
| Zero-Noise Extrapolation (ZNE) | Noise amplification and extrapolation | Moderate (2-10x) | General optimization problems [1] |
| Symmetry Verification | Post-selection based on symmetry | Problem-dependent | Quantum chemistry (particle number conservation) [1] |
| Probabilistic Error Cancellation | Quasi-probability decomposition | High (exponential in error rates) | Small circuits with low error rates [1] |
| Readout Error Mitigation | Measurement error calibration | Exponential in number of qubits | All measurement-intensive applications [1] |
Table 3: Essential Research Reagents and Computational Resources
| Resource Category | Specific Examples | Function/Role | Implementation Notes |
|---|---|---|---|
| Quantum Hardware Platforms | Superconducting qubits (IBM, Google), Trapped ions (Quantinuum) | Physical implementation of quantum circuits | 50-1000 qubits; coherence times ~100μs; gate fidelities 95-99.9% [2] [1] |
| Classical Optimizers | COBYLA, SPSA, Adam, BFGS | Adjust quantum circuit parameters to minimize energy | Choice affects convergence speed and resilience to noise [2] |
| Quantum Development Frameworks | Qiskit (IBM), Cirq (Google), Pennylane (Xanadu) | Algorithm design, circuit compilation, and execution | Enable hybrid quantum-classical programming models [7] |
| Error Mitigation Tools | ZNE, symmetry verification, PEC | Reduce impact of noise without quantum error correction | Critical for achieving chemical accuracy on NISQ devices [1] |
| Chemical Computation Libraries | OpenFermion, PySCF, QChemistry | Molecular Hamiltonian generation and classical comparison | Provide electronic structure baselines and problem encoding [4] |
Current Challenges and Research Frontiers
Fundamental Limitations
Despite promising developments, VQAs face significant challenges that must be addressed to achieve practical quantum advantage in combinatorial chemistry:
Barren Plateaus: The gradient of the cost function can vanish exponentially with system size, making optimization practically impossible for large molecules [2] [8]. Current research explores adaptive ansätze and informed initialization to mitigate this issue [2].
Ansatz Expressibility and Depth: Trade-offs exist between chemical accuracy (requiring expressive ansätze like UCCSD) and NISQ feasibility (favoring hardware-efficient designs with lower depth) [2].
Measurement Overhead: The number of measurements required to achieve chemical accuracy scales polynomially with system size, creating a significant bottleneck for larger molecules [2].
Optimization Complexity: Training VQA parameters has been shown to be NP-hard in general, suggesting that optimal parameter discovery may be as difficult as solving the original problem [6].
Emerging Solutions and Future Directions
Research continues to address these limitations through several promising avenues:
- Problem Decomposition: Fragment molecular orbital (FMO) approaches combined with VQE show potential for simulating larger molecular systems by partitioning into manageable fragments [1].
- Parallelization Strategies: Recent frameworks enable splitting quantum circuits to solve problems larger than available qubits, approximating the ground state distribution through smaller, executable circuits [6].
- Advanced Ansatz Designs: Adaptive ansätze that balance chemical intuition with hardware constraints are under active development to improve convergence and accuracy [2].
- Error-Aware Optimization: Optimizers specifically designed to function effectively in noisy environments are being developed to enhance algorithmic resilience [2].
Variational Quantum Algorithms represent the most viable pathway toward practical quantum-enhanced computational chemistry in the NISQ era. While significant challenges remain in scaling, optimization, and error mitigation, the continued development of VQE and its variants has established a foundation for potentially transformative advances in molecular simulation. The hybrid quantum-classical framework of VQAs provides combinatorial chemistry researchers with an accessible entry point to quantum computational methods while accommodating current hardware limitations. As quantum processors continue to improve in scale and fidelity, and algorithmic innovations address existing limitations, VQAs are positioned to play an increasingly important role in accelerating drug discovery and materials design.
Variational Quantum Algorithms (VQAs) represent a leading paradigm for harnessing the computational potential of current noisy intermediate-scale quantum (NISQ) devices [9] [10]. These hybrid quantum-classical algorithms leverage a cooperative process where a quantum computer prepares and measures parameterized quantum states, while a classical computer optimizes those parameters to find approximate solutions to complex problems [6]. For researchers in combinatorial chemistry and drug development, VQAs offer a promising path to simulating molecular systems and optimizing molecular structures, tasks that are often intractable for classical computers alone [10] [11]. The efficacy of any VQA rests on three fundamental pillars: the cost function, which defines the problem's objective; the parameterized quantum circuit (ansatz), which defines the search space; and the classical optimizer, which navigates that space to find the optimal solution [12] [13]. This guide provides an in-depth technical examination of these core components, contextualized for applications in quantum chemistry.
The Variational Quantum Algorithm Workflow
At its core, a VQA is a hybrid feedback loop [10] [12]. The quantum computer executes a parameterized circuit (the ansatz) to prepare a quantum state. This state is measured to evaluate a cost function, which encodes the problem of interest—such as the energy of a molecule. The classical optimizer then analyzes this cost value and updates the circuit parameters for the next iteration. This process repeats until the cost function converges, signaling that an approximate solution has been found [12].
The following diagram illustrates this hybrid workflow and the interplay between its core components.
Component 1: Cost Functions
The cost function, ( C(\theta) ), is the objective function that the VQA seeks to minimize. It provides a quantitative measure of how "good" the current parameterized quantum state is for solving the target problem [12] [13]. A general and task-agnostic form of the cost function can be expressed as: [ C(\bm{\theta}) = f\left[\mathcal{U}^{\dagger}(\bm{\theta}), \mathcal{U}(\bm{\theta}), {\rhok}, {Ok}\right] ] for a super function ( f ), where ( {\rhok} ) is a set of input quantum states and ( {Ok} ) is a set of Hermitian observables or measurement operators [9].
Cost Functions in Quantum Chemistry
In the context of quantum chemistry, the most prominent cost function is the expectation value of a molecular electronic structure Hamiltonian, ( H ). This is the foundation of the Variational Quantum Eigensolver (VQE) [10] [11]. The cost function is given by: [ C(\theta) = \langle \psi(\theta) | H | \psi(\theta) \rangle ] where ( |\psi(\theta)\rangle ) is the quantum state generated by the ansatz. The variational principle of quantum mechanics guarantees that ( C(\theta) \geq E0 ), where ( E0 ) is the true ground-state energy [12]. Minimizing ( C(\theta) ) thus yields an upper-bound approximation of ( E_0 ), which is fundamental for understanding molecular stability, reaction pathways, and other chemical properties [12].
Table: Key Cost Functions and Their Applications in Chemistry
| Cost Function | Mathematical Formulation | Primary Application in Chemistry | ||||
|---|---|---|---|---|---|---|
| Energy Expectation [12] | ( C(\theta) = \langle 0 | U^\dagger(\theta) H U(\theta) | 0 \rangle ) | Finding molecular ground-state energies (VQE). | ||
| Hilbert-Schmidt Distance [9] | ( C(\theta) = 1 - \frac{1}{2^n} \left | \text{Tr}( \mathcal{U}^\dagger(\theta) \mathcal{V} ) \right | ^2 ) | Variational Quantum Compilation (VQC) for quantum simulation. | ||
| Fidelity with Target State | ( C(\theta) = 1 - \left | \langle \psi_{\text{target}} | U(\theta) | 0 \rangle \right | ^2 ) | Preparing specific molecular wavefunctions or trial states. |
Component 2: Parameterized Quantum Circuits (Ansätze)
The parameterized quantum circuit (PQC), or ansatz, ( U(\theta) ), is the function that maps a set of classical parameters ( \theta ) to a region of the Hilbert space [13]. The structure of the ansatz critically determines the expressivity of the model and the efficiency of the optimization [12] [14]. A common construction is a sequence of quantum gates: [ \mathcal{U}(\bm{\theta}) = UM(\thetaM) \cdots U2(\theta2)U1(\theta1) ] where individual gates are often parameterized rotations, e.g., ( Uj(\thetaj) = \exp(-i\thetaj Pj/2) ) and ( P_j ) is a Pauli matrix [9].
Ansatz Strategies for Chemical Problems
Choosing an ansatz is a non-trivial task that balances expressivity, trainability, and hardware efficiency. For chemical applications, the two primary approaches are physically-inspired and hardware-efficient ansätze.
- Physically-Inspired Ansätze: These, such as the Unitary Coupled Cluster (UCC) ansatz, are designed to efficiently encode the structure of molecular Hamiltonians. They are known to generate chemically meaningful states and are often very accurate, but can lead to deep quantum circuits that are challenging for NISQ devices [10].
- Hardware-Efficient Ansätze (HEA): These ansätze are constructed from native gates specific to a particular quantum processor. They typically consist of alternating layers of single-qubit rotations and entangling gates, creating shallow circuits that are more resilient to noise [6]. The trade-off is that their structure may not be tailored to the chemical problem, which can lead to harder optimization landscapes, including issues like Barren Plateaus [11].
Component 3: Classical Optimizers
The classical optimizer is the engine that drives the parameter update rule, ( \theta \to \theta' ), to minimize the cost function [15] [13]. The choice of optimizer is critical for the convergence speed and final result quality of a VQA, especially given the noisy and expensive nature of quantum evaluations.
Gradient-Based and Gradient-Free Methods
Optimizers for VQAs can be broadly categorized based on their use of gradient information.
- Gradient-Based Optimizers: These methods use information about the gradient of the cost function, ( \nabla\theta C(\theta) ), to inform parameter updates. A cornerstone technique is the parameter-shift rule, which provides an exact gradient for certain circuits by evaluating the cost function at two different points [9]. For a parameter ( \thetaj ) of a gate ( Uj(\thetaj) = \exp(-i\thetaj Pj/2) ), the partial derivative is given by: [ \frac{\partial C(\bm{\theta})}{\partial \thetaj} = \frac{1}{2}\left[ C(\bm{\theta} + \frac{\pi}{2}\bm{e}j) - C(\bm{\theta} - \frac{\pi}{2}\bm{e}_j) \right] ] This rule allows for analytic gradient calculation on quantum hardware and is more accurate and stable than finite-difference methods [9].
- Gradient-Free Optimizers: Methods such as COBYLA, Nelder-Mead, and SPSA do not require gradient information and are often used when the parameter-shift rule is not applicable or when the cost function evaluations are too noisy [15]. While more robust to noise, they typically require more function evaluations to converge.
The diagram below illustrates the quantum circuit and measurement process required for computing gradients using the parameter-shift rule.
Table: Comparison of Classical Optimizers for VQAs
| Optimizer Type | Examples | Key Characteristics | Suitability for Chemistry VQAs |
|---|---|---|---|
| Gradient-Based | Adam, SPSA,Parameter-Shift [9] [15] | Uses gradient info for faster convergence; can be sensitive to noise. | Ideal for high-precision energy calculations when exact gradients are available via parameter-shift. |
| Gradient-Free | COBYLA, Nelder-Mead [15] | Robust to noise and stochastic cost function evaluations; slower convergence. | Useful for initial ansatz training or on extremely noisy hardware where gradient estimation is unreliable. |
| Noise-Aware | Novel methods from"noise-aware" optimization literature [15] | Explicitly models and accounts for noise in the cost function. | Promising for obtaining more reliable results from current quantum hardware without extra error mitigation. |
An Integrated View: The Scientist's Toolkit for VQA Experiments
Successfully deploying a VQA for a chemistry problem requires the careful selection and integration of all three components. The following toolkit outlines the essential "research reagents" and their roles in a typical experiment, such as calculating the ground state energy of a molecule.
Table: Essential "Research Reagents" for a VQA Chemistry Experiment
| Tool Category | Specific Choice | Function in the Experiment |
|---|---|---|
| Problem Encoding | Jordan-Wigner / Bravyi-Kitaev Transformation | Maps the molecular fermionic Hamiltonian to a qubit Hamiltonian (e.g., an Ising model). |
| Ansatz Circuit | Unitary Coupled Cluster (UCCSD) / Hardware-Efficient Ansatz | Defines the space of quantum states in which to search for the molecular ground state. |
| Cost Function | Energy Expectation Value ( \langle H \rangle ) | Encodes the target molecular property (ground state energy) as a minimizable function. |
| Gradient Method | Parameter-Shift Rule [9] | Enables efficient and exact calculation of gradients for parameter updates directly from quantum measurements. |
| Classical Optimizer | BFGS / Adam / COBYLA | The classical routine that iteratively updates the quantum circuit parameters to minimize the energy. |
Experimental Protocol: Variational Quantum Eigensolver (VQE) for Ground State Energy
This protocol details the steps for a typical VQE experiment aimed at finding the ground state energy of a molecule.
- Problem Formulation: Begin with the molecular geometry and compute the second-quantized electronic Hamiltonian using a classical quantum chemistry package. Transform this Hamiltonian into a qubit-representable form (( H )) using a mapping like Jordan-Wigner or Bravyi-Kitaev.
- Ansatz Initialization: Select an ansatz ( U(\theta) ) (e.g., UCCSD or HEA) and initialize its parameters ( \theta ). Initialization can be random, or based on classical heuristics for faster convergence.
- Quantum Processing: For the current set of parameters ( \theta ): a. Prepare the initial state ( |0\rangle^{\otimes n} ). b. Apply the parameterized circuit ( U(\theta) ). c. Measure the expectation values of the individual Pauli terms that constitute ( H ). This often requires repeating the state preparation and measurement many times to achieve sufficient statistical accuracy.
- Classical Processing: a. Cost Calculation: Classically combine the measured expectation values to compute the total energy ( C(\theta) = \langle H \rangle ). b. Optimization: Feed the cost value (and gradients, if using a gradient-based method) to the classical optimizer. c. Parameter Update: The optimizer proposes a new set of parameters ( \theta' ).
- Iteration and Convergence: Steps 3 and 4 are repeated until the energy ( C(\theta) ) converges within a predefined threshold or a maximum number of iterations is reached. The final value of ( C(\theta) ) is the approximation of the ground-state energy.
Advanced Concepts and Current Research
The field of VQAs is rapidly evolving to address significant challenges and expand applications.
- Barren Plateaus: A key challenge is the barren plateau phenomenon, where the gradients of the cost function vanish exponentially with the number of qubits, making optimization practically impossible [11]. Research into problem-inspired ansätze and smart initialization strategies is ongoing to mitigate this issue [11].
- Scalability and Parallelization: To overcome hardware limitations on qubit count, frameworks for parallelizing VQAs have been proposed [6]. These methods split a large quantum circuit into smaller, independent sub-circuits (slices) that can be executed in parallel on a quantum device with fewer qubits. A global objective function then guides the optimization of these parallel slices, allowing for the solution of problems larger than the physical qubit count [6].
- Expressivity and Universality: Theoretical work has established that parameterized quantum circuits can serve as universal generative models for continuous multivariate distributions [14]. This has implications for modeling complex molecular properties. The expressivity, however, involves a trade-off: generating high-dimensional distributions requires either a large number of qubits or a large number of measurements with high-precision observables [14].
The synergistic operation of cost functions, parameterized circuits, and classical optimizers forms the foundation of all Variational Quantum Algorithms. For the combinatorial chemistry researcher, a deep understanding of these components—from selecting a chemically meaningful cost function and an appropriate, trainable ansatz, to choosing a robust classical optimizer—is essential for leveraging NISQ-era quantum computers effectively. While challenges such as barren plateaus and hardware noise remain active areas of research, the continued development of VQAs provides a tangible and promising path toward achieving quantum utility in simulating and designing molecular systems.
Variational Quantum Algorithms (VQAs) represent a class of hybrid quantum-classical methods designed to leverage the capabilities of current noisy intermediate-scale quantum (NISQ) devices. These algorithms combine quantum state preparation and measurement with classical optimization, making them particularly suitable for the constrained quantum hardware of today. Within the domain of combinatorial chemistry research, two algorithms stand out for their potential: the Variational Quantum Eigensolver (VQE) and the Quantum Approximate Optimization Algorithm (QAOA). VQE is primarily used to find the ground state energy of molecular systems, a fundamental problem in quantum chemistry and drug discovery [16] [17] [18]. QAOA, while applicable to broader combinatorial optimization problems, can be adapted for specific chemical simulations, such as protein folding and molecular conformation analysis [17]. This technical guide provides an in-depth analysis of both algorithms, detailing their theoretical foundations, methodological protocols, and practical applications in chemical research.
The Variational Quantum Eigensolver (VQE)
Theoretical Foundation
The Variational Quantum Eigensolver is a hybrid quantum-classical algorithm that applies the variational principle of quantum mechanics to find the ground state energy of a given physical system, typically a molecular Hamiltonian. The core principle relies on the fact that the expectation value of a Hamiltonian ( \hat{H} ) in any trial state ( |\psi(\vec{\theta})\rangle ) is always greater than or equal to the true ground state energy ( E_0 ):
[ E(\vec{\theta}) = \langle \psi(\vec{\theta}) | \hat{H} | \psi(\vec{\theta}) \rangle \geq E_0 ]
The algorithm proceeds by preparing a parameterized quantum circuit (ansatz) on a quantum processor, measuring the expectation value of the Hamiltonian, and using a classical optimizer to adjust the parameters ( \vec{\theta} ) to minimize this expectation value [16] [19]. This process iteratively converges toward an approximation of the ground state energy.
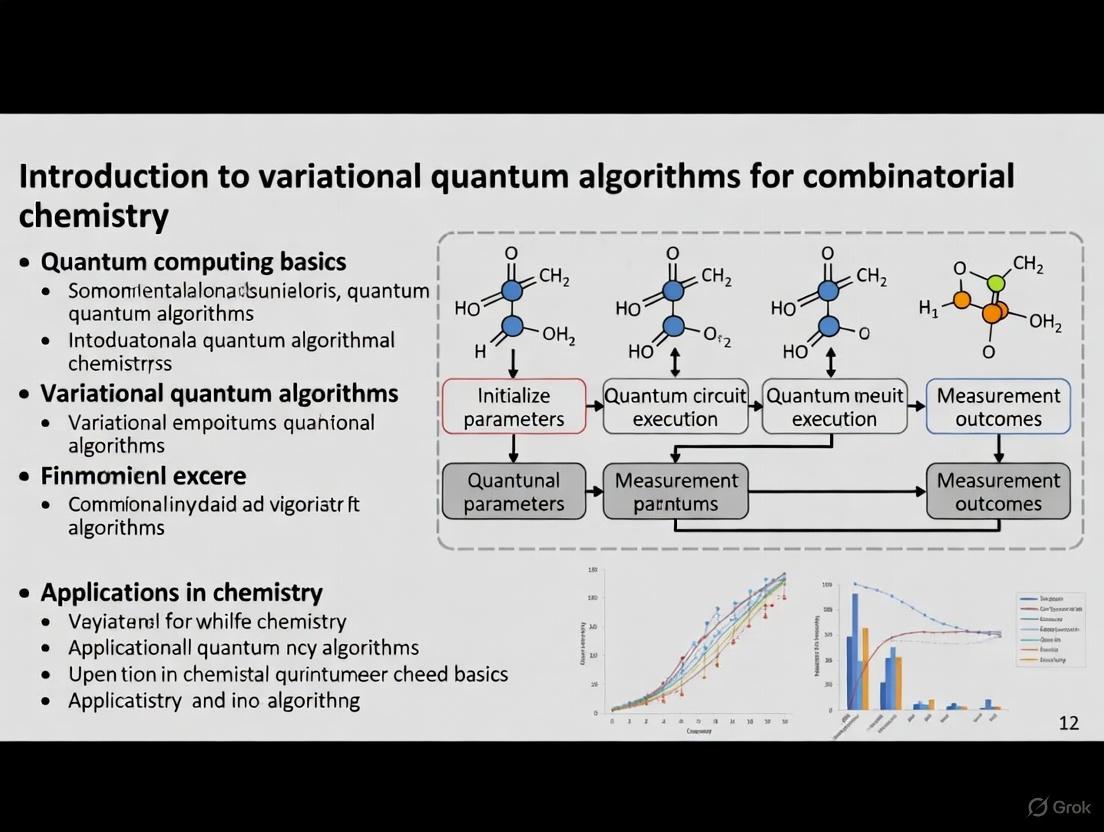
Algorithmic Workflow and Protocol
The standard VQE protocol involves several key steps, as visualized in the workflow diagram below.
Step-by-Step Protocol:
Hamiltonian Formulation: The first step involves defining the electronic Hamiltonian of the target molecule in the second quantized form and then mapping it to a qubit representation using a fermion-to-qubit transformation such as the Jordan-Wigner or Bravyi-Kitaev transformation [16] [19]. For a hydrogen molecule (( \mathrm{H}_2 )), this results in a Hamiltonian composed of a sum of Pauli terms:
H = -0.0996*I + 0.1711*Z(0) + 0.1711*Z(1) + 0.1686*(Z(0)@Z(1)) + ...[19].Ansatz Selection: A parametrized quantum circuit (the ansatz) is chosen to prepare the trial wavefunction. Common choices in quantum chemistry include the Unitary Coupled Cluster (UCC) ansatz, which captures electronic excitations. A simpler, hardware-efficient ansatz is often used to reduce circuit depth on NISQ devices [16]. For the ( \mathrm{H}_2 ) example, a circuit initializes the Hartree-Fock state
|1100⟩and applies aDoubleExcitationgate to introduce correlation [19].Measurement and Expectation Estimation: The expectation value of the Hamiltonian is calculated by measuring each Pauli term in the decomposed Hamiltonian. This often requires running the circuit multiple times to obtain sufficient statistical accuracy [16].
Classical Optimization: A classical optimizer (e.g., gradient descent, SPSA) processes the measured energy and computes updates for the circuit parameters ( \vec{\theta} ). This step is repeated until the energy converges to a minimum [16] [19].
Key Components for VQE Experiments
Table 1: Essential "Research Reagent Solutions" for a VQE Experiment
| Component Name | Type/Function | Role in the VQE Protocol |
|---|---|---|
| Molecular Hamiltonian | Mathematical Model | Encodes the physical system and problem; the target observable whose ground state is sought [19]. |
| Qubit Encoding (e.g., Jordan-Wigner) | Mapping Technique | Transforms the fermionic Hamiltonian of a molecule into a form (Pauli strings) executable on a qubit-based quantum processor [16] [19]. |
| Parametrized Ansatz (e.g., UCC) | Quantum Circuit | Generates the trial wavefunction ( |\psi(\vec{\theta})\rangle ); its expressibility determines how closely the true ground state can be approximated [16]. |
| Classical Optimizer (e.g., Gradient Descent) | Classical Algorithm | Adjusts the parameters ( \vec{\theta} ) of the quantum circuit to minimize the expectation value of the Hamiltonian [16] [19]. |
The Quantum Approximate Optimization Algorithm (QAOA)
Theoretical Foundation
The Quantum Approximate Optimization Algorithm is a variational algorithm designed to find approximate solutions to combinatorial optimization problems. While its application to direct quantum chemistry is less straightforward than VQE, it is highly relevant for problems like protein folding and resource allocation in drug discovery [17] [20]. QAOA solves problems by encoding the solution to a cost Hamiltonian ( H_C ), whose ground state corresponds to the optimal solution.
The algorithm constructs a variational ansatz by alternately applying the cost Hamiltonian and a mixer Hamiltonian ( HM ) (typically a sum of Pauli-X operators) for ( p ) layers. The quantum state is prepared as: [ |\psi(\boldsymbol{\gamma}, \boldsymbol{\beta})\rangle = \prod{k=1}^{p} e^{-i\betak HM} e^{-i\gammak HC} |+\rangle^{\otimes n} ] where ( \boldsymbol{\gamma} ) and ( \boldsymbol{\beta} ) are variational parameters optimized to minimize ( \langle H_C \rangle ) [21] [22].
Algorithmic Workflow and Protocol
The following diagram illustrates the iterative QAOA workflow for solving combinatorial problems.
Step-by-Step Protocol:
Problem Encoding: The combinatorial problem (e.g., Minimum Vertex Cover, MaxCut, or a molecular similarity search) is mapped to a cost Hamiltonian ( HC ). For a graph problem, ( HC ) might be: ( HC = \sum{(i,j) \in E} w{ij} Zi Z_j ), where the ground state corresponds to the solution [21] [22].
Circuit Construction: The quantum circuit is built with ( p ) layers. Each layer consists of a cost unitary ( e^{-i\gammak HC} ) and a mixer unitary ( e^{-i\betak HM} ). The initial state is the uniform superposition state ( |+\rangle^{\otimes n} ) [22].
Measurement and Classical Optimization: The circuit is executed, and the expectation value ( \langle H_C \rangle ) is measured. A classical optimizer adjusts the parameters ( \boldsymbol{\gamma}, \boldsymbol{\beta} ) to minimize this expectation. After optimization, the final circuit is sampled multiple times to obtain a distribution of candidate solutions, with the most frequent bitstrings representing high-quality approximate answers [23] [22].
Key Components for QAOA Experiments
Table 2: Essential "Research Reagent Solutions" for a QAOA Experiment
| Component Name | Type/Function | Role in the QAOA Protocol |
|---|---|---|
| Cost Hamiltonian (H_C) | Mathematical Model | Encodes the objective function of the combinatorial optimization problem to be solved [22]. |
| Mixer Hamiltonian (H_M) | Quantum Operator | Facilitates transitions between classical states (e.g., ( \sumi Xi )), ensuring exploration of the solution space [21] [22]. |
| Parameterized Quantum Circuit | Quantum Circuit | The QAOA ansatz, structured by depth ( p ), which applies alternating layers of cost and mixer unitaries [22]. |
| Classical Optimizer | Classical Algorithm | Finds optimal parameters ( \boldsymbol{\gamma}, \boldsymbol{\beta} ) that minimize the expectation value of ( H_C ) [22]. |
Comparative Analysis and Applications in Chemistry
Algorithm Comparison
Table 3: Comparative Analysis of VQE and QAOA
| Feature | Variational Quantum Eigensolver (VQE) | Quantum Approximate Optimization Algorithm (QAOA) |
|---|---|---|
| Primary Application | Quantum Chemistry (Ground State Energy) [16] [19] | Combinatorial Optimization (e.g., Protein Folding) [17] [20] |
| Core Principle | Quantum Variational Principle [16] | Alternating Operator Ansatz [21] |
| Problem Encoding | Molecular Hamiltonian (via Jordan-Wigner, etc.) [16] | Cost Hamiltonian (e.g., Ising Model) [22] |
| Ansatz Structure | Chemistry-inspired (UCC) or Hardware-efficient [16] | Fixed structure with cost/mixer layers of depth ( p ) [22] |
| Key Advantage | Directly applicable to molecules; more robust to noise [16] | Natural for combinatorial problems; well-defined circuit structure [23] |
| Key Challenge | High-dimensional parameter optimization; barren plateaus [16] [24] | Parameter optimization difficulty; performance tied to depth ( p ) [23] |
Applications in Chemical Research and Drug Discovery
VQE has been successfully demonstrated on small molecules like hydrogen (( \mathrm{H}_2 )), where it can converge to the full configuration interaction (FCI) energy, achieving results like -1.13726250 Ha, which is very close to the FCI benchmark [19]. Research is ongoing to scale these simulations to larger, more chemically relevant molecules like lithium-based compounds or catalysts [18] [24].
Both VQE and QAOA show promise for tackling the complex problem of protein folding. This involves finding the stable, three-dimensional structure of a protein from its amino acid sequence—a monumental combinatorial optimization task [17]. Early research indicates that quantum algorithms may explore the vast conformational space of proteins more efficiently than classical methods, potentially accelerating drug discovery pipelines.
Advanced Techniques and Future Outlook
Recent advancements are pushing the boundaries of these core algorithms. ADAPT-QAOA constructs the ansatz iteratively, adding operators that provide the largest energy gradient, leading to more compact and powerful circuits [21]. Furthermore, machine learning techniques are being integrated to address optimization challenges. The QAOA-GPT framework uses generative pre-trained transformers to predict optimized quantum circuits and their parameters for given problem instances, effectively bypassing the expensive classical optimization loop [21].
For VQE, ongoing research focuses on developing better ansätze, more efficient measurement techniques to reduce the number of circuit repetitions, and advanced error mitigation strategies to combat the effects of noise on NISQ hardware [24]. As quantum hardware continues to scale and noise levels are reduced, VQE and QAOA are poised to play a pivotal role in achieving a quantum advantage in computational chemistry and drug development.
Quantum computational chemistry holds the profound promise of simulating molecular systems with an accuracy that is intractable for classical computers [25]. Everything in chemistry—from bonds and reactions to catalysts and materials—stems from the quantum behavior of electrons. Classical computing methods, including density functional theory, often struggle to calculate the behavior of strongly correlated electrons and must rely on approximations that are not always completely accurate [25]. Quantum computers, by contrast, are natural quantum systems themselves. They can, in principle, determine the exact quantum state of all electrons and compute their energy and molecular structures without these approximations because, like qubits, electrons exist in multiple states at once [25]. This capability could fundamentally revolutionize the design of new drugs, materials, and catalysts.
This technical guide details the process of mapping complex chemical problems onto quantum hardware, framing the discussion within the broader thesis of employing variational quantum algorithms for combinatorial chemistry research. It is designed for researchers, scientists, and drug development professionals who seek to understand and implement these cutting-edge techniques. We will explore the foundational principles, the current state of experimental protocols, and the practical tools required to embark on this transformative research path.
Theoretical Foundations: From Molecules to Qubits
The Electronic Structure Problem
At the heart of quantum computational chemistry is the electronic structure problem. The goal is to solve the time-independent Schrödinger equation for a molecule:
$$\hat{H}|\Psi\rangle = E|\Psi\rangle$$
Here, $\hat{H}$ is the molecular Hamiltonian, the operator corresponding to the total energy of the system. $|\Psi\rangle$ is the wavefunction, a mathematical description of the quantum state of the molecule's electrons, and $E$ is the total energy associated with that state [26]. Solving this equation unlocks all chemical properties, but the complexity of $\hat{H}$ grows exponentially with the number of electrons, making exact solutions for all but the smallest molecules impossible for classical computers.
The Mapping Process
To solve this problem on a quantum computer, a multi-step mapping process is required to translate the physical molecular system into a form executable on quantum hardware.
1. Molecular Hamiltonian Formulation: The first step is to define the molecular Hamiltonian in the second quantized form, which describes the system in terms of creation and annihilation operators acting on a reference state (e.g., the Hartree-Fock state) [26]. This Hamiltonian encompasses the kinetic energies of the electrons and the potential energies from electron-nuclei and electron-electron interactions.
2. Qubit Encoding: The next critical step is to encode the fermionic Hamiltonian into a qubit Hamiltonian. This is necessary because the creation and annihilation operators of electrons obey fermionic anti-commutation relations, while the Pauli operators governing qubits obey different algebraic rules. Common encoding methods include:
- Jordan-Wigner Transformation: Maps fermionic operators to strings of Pauli operators ($X$, $Y$, $Z$). It is straightforward but can result in long Pauli strings, increasing circuit depth.
- Bravyi-Kitaev Transformation: A more sophisticated mapping that offers a logarithmic scaling of Pauli string length for some operations, often providing a more efficient representation [26].
After encoding, the molecular Hamiltonian is expressed as a linear combination of Pauli strings:
$$\hat{H} = \sumi ci P_i$$
where $Pi$ are Pauli strings (e.g., $X0 Z1 Y3$) and $c_i$ are real coefficients.
3. Ansatz Selection: The encoded Hamiltonian is now ready for a quantum computer. A parameterized quantum circuit, or ansatz, is chosen to prepare a trial wavefunction $|\Psi(\vec{\theta})\rangle$. The parameters $\vec{\theta}$ are then variationally optimized to find the lowest energy eigenvalue, which corresponds to the molecule's ground state. The choice of ansatz is pivotal, balancing expressiveness with computational feasibility on noisy hardware.
The following diagram illustrates this core mapping workflow from a chemical system to a quantum computation.
Key Quantum Algorithms and Experimental Protocols
The Variational Quantum Eigensolver (VQE)
The Variational Quantum Eigensolver (VQE) is a leading hybrid quantum-classical algorithm designed to find the ground state energy of a molecule, making it a cornerstone for quantum computational chemistry [27]. It is particularly well-suited for the current Noisy Intermediate-Scale Quantum (NISQ) era because it does not require long, fault-tolerant quantum circuits.
The VQE protocol follows these steps [26] [27]:
- Problem Mapping: The molecular Hamiltonian is mapped to a qubit Hamiltonian as described in Section 2.2.
- Quantum Subroutine: A parameterized quantum circuit (ansatz) prepares a trial state $|\Psi(\vec{\theta})\rangle$ on the quantum processor.
- Measurement: The quantum computer measures the expectation value $\langle \Psi(\vec{\theta}) | \hat{H} | \Psi(\vec{\theta}) \rangle$. This is typically done by measuring the expectation value of each Pauli term $P_i$ in the Hamiltonian and then combining the results classically.
- Classical Optimization: A classical optimizer (e.g., COBYLA, SPSA) receives the measured energy and adjusts the parameters $\vec{\theta}$ to minimize the energy.
- Iteration: Steps 2-4 are repeated until the energy converges to a minimum, which is reported as the approximation of the ground state energy.
Advanced Hybrid Methodologies
Recent research has demonstrated more sophisticated hybrid workflows that leverage the strengths of both quantum and classical high-performance computing (HPC). A landmark study from Caltech, IBM, and RIKEN provides a powerful template [28].
Protocol: Quantum-Centric Supercomputing for Complex Molecules
- Objective: To determine the electronic energy levels of an iron-sulfur cluster ([4Fe-4S]), a system biologically crucial for nitrogen fixation and notoriously challenging for classical algorithms [28].
- Methodology:
- Quantum Pre-processing: An IBM quantum device (Heron processor) was used to simplify the problem. Instead of relying on classical heuristics to identify the most important components of the massive Hamiltonian matrix, the quantum computer rigorously identified the most relevant components.
- Classical Post-processing: The reduced, relevant part of the Hamiltonian was then fed into the RIKEN Fugaku supercomputer to solve for the exact wavefunction and energy.
- Key Insight: This workflow demonstrates that quantum computers can act as specialized co-processors, not necessarily to solve the entire problem, but to pre-process and simplify intractable problems for classical HPC systems [28].
The diagram below visualizes this advanced hybrid workflow.
Quantum Machine Learning Enhancements
To overcome limitations in accuracy due to hardware noise and shallow circuit depths, researchers are integrating neural networks with quantum algorithms. The hybrid quantum-neural wavefunction (pUNN) is one such innovation [26].
- Principle: This framework uses a shallow quantum circuit (e.g., a paired Unitary Coupled-Cluster with double excitations, pUCCD) to learn the quantum phase structure, which is difficult for neural networks alone. A classical neural network then corrects the amplitude of the wavefunction to account for contributions outside the simplified model's subspace [26].
- Protocol: The quantum circuit and neural network are trained jointly. An efficient measurement algorithm avoids the costly need for full quantum state tomography.
- Experimental Validation: This method was successfully validated on a superconducting quantum computer by computing the reaction barrier for the isomerization of cyclobutadiene, a challenging multi-reference model, achieving high accuracy and significant noise resilience [26].
Hardware Landscape and Resource Estimation
The practical implementation of these algorithms depends critically on the underlying quantum hardware. Current hardware is rapidly evolving, with different qubit modalities offering distinct advantages.
Table 1: Key Quantum Hardware Platforms and Performance Metrics (2025)
| Company | Qubit Modality | Recent Processor | Key Performance Metrics | Roadmap Highlights |
|---|---|---|---|---|
| IBM [29] [30] | Superconducting | Nighthawk (120 qubits), Heron r3 | Heron r3: 57 two-qubit couplings with error rate <0.001; 330,000 CLOPS | 200 logical qubits by 2029; Quantum-centric supercomputers with 100,000 qubits by 2033 |
| Google [31] [30] | Superconducting | Willow (105 qubits) | Single-qubit gate fidelity: 99.97%; Entangling gate fidelity: 99.88% | Useful, error-corrected quantum computer by 2029 |
| IonQ [32] [30] | Trapped Ions | 36-qubit system | Outperformed classical HPC by 12% in a medical device simulation | Broad quantum advantage targeted by 2025; Rack-mounted systems for data centers |
| Microsoft/Atom Computing [32] [30] | Topological / Neutral Atoms | Majorana 1 / Neutral Atom platform | Demonstrated 28 logical qubits; 1000-fold error reduction | Scaling to 1,000+ qubit systems by 2026; Focus on inherent qubit stability |
The resources required to solve industrially relevant chemistry problems are substantial. Early estimates suggested that simulating complex metalloenzymes like cytochrome P450 or the iron-molybdenum cofactor (FeMoco) would require millions of physical qubits [25]. However, recent breakthroughs in error correction and algorithmic efficiency are rapidly reducing these requirements. For example, the French start-up Alice & Bob has estimated that its qubit design could reduce the qubit count for simulating FeMoco to just under 100,000 [25]. Progress in error correction, such as Google's demonstration of "below-threshold" operation and IBM's development of fast decoders, is critical to bridging this gap [32] [29].
Table 2: Representative Chemistry Problems and Algorithmic Applications
| Target System | Chemical Significance | Quantum Algorithm(s) Applied | Reported Outcome / Milestone |
|---|---|---|---|
| Iron-Sulfur Cluster [4Fe-4S] [28] | Essential for biological nitrogen fixation and electron transfer. | Hybrid quantum-classical (IBM-Caltech) | Used 77 qubits to simplify Hamiltonian for classical solution; a step beyond exact diagonalization limits. |
| Cyclobutadiene [26] | A challenging model for pericyclic reactions and multi-reference character. | pUNN (Hybrid quantum-neural) | Achieved high accuracy and noise resilience for isomerization barrier on a superconducting quantum processor. |
| Nitrogen Fixation [25] | Critical for fertilizer production and understanding biological catalysts. | Enhanced VQE (Qunova Computing) | Modeled nitrogen reactions almost 9x faster than classical methods in tests. |
| Cytochrome P450 [32] | Key human enzyme involved in drug metabolism. | Quantum Simulation (Google & Boehringer Ingelheim) | Simulated with greater efficiency and precision than traditional methods. |
| Small Molecules (H₂, LiH, BeH₂) [25] | Benchmark systems for validating quantum algorithms. | Variational Quantum Eigensolver (VQE) | Successfully estimated ground-state energy, proving fundamental principles. |
To conduct research in this field, a combination of quantum hardware access, classical software, and theoretical knowledge is required. The following table details key "research reagents" for the modern quantum computational chemist.
Table 3: Essential Research Reagents and Resources for Quantum Computational Chemistry
| Resource Category | Item / Platform | Function and Relevance |
|---|---|---|
| Quantum Hardware Access | IBM Quantum Systems [29], Google Quantum AI [31], IonQ Forte [32] | Provides cloud-based access (QaaS) to real quantum processors for running experiments and algorithms. Essential for empirical validation. |
| Software Development Kits (SDKs) | Qiskit [29] | An open-source SDK for quantum computing. Used to define quantum circuits, execute them on simulators or hardware, and implement error mitigation. |
| Algorithmic Primitives | Variational Quantum Eigensolver (VQE) [27], Quantum Approximate Optimization Algorithm (QAOA) [27] | Core algorithmic frameworks for solving chemistry and optimization problems on NISQ-era hardware. |
| Advanced Software Tools | Qiskit Functions [29], Samplomatic [29] | Pre-built functions for specific chemistry problems and tools for implementing advanced, composable error mitigation techniques. |
| Classical Computing Resources | High-Performance Computing (HPC) Clusters [28] | Essential for running the classical optimization loops in VQE, pre-/post-processing data, and hybrid workflows like quantum-centric supercomputing. |
| Theoretical Frameworks | Unitary Coupled Cluster (UCC) [26], Quantum Machine Learning (QML) [26] [27] | Provides the mathematical ansatz for preparing trial wavefunctions and advanced methods for enhancing accuracy and resilience. |
| Error Mitigation Techniques | Probabilistic Error Cancellation (PEC) [29], Dynamic Circuits [29] | A set of methods to reduce the impact of noise on quantum计算结果, crucial for obtaining meaningful results from current imperfect hardware. |
The path from a molecular Hamiltonian to a computation on a qubit-based processor is now a well-defined, though rapidly evolving, discipline. Framed by the power of variational quantum algorithms, researchers are steadily progressing from simulating small benchmark molecules to tackling systems of direct industrial and scientific importance, such as the iron-sulfur clusters pivotal in biochemistry [28]. While significant challenges in hardware scalability and error correction remain, the synergistic development of more robust algorithms like VQE and pUNN, coupled with advanced hybrid methodologies, is providing a clear and practical roadmap. For researchers in combinatorial chemistry and drug development, engaging with these tools and protocols today is no longer speculative but a strategic step towards leveraging a transformative technology that promises to redefine the limits of molecular simulation.
The Promise of Quantum Advantage in Simulating Quantum Systems
The simulation of quantum systems represents a foundational application where quantum computers are poised to deliver significant advantage over classical methods. Within combinatorial chemistry research, the challenge of determining molecular electronic structures and dynamics is fundamentally constrained by the exponential scaling of complexity on classical computers. Quantum computers, which operate using the same principles as the molecular systems being simulated, offer a path to overcome this bottleneck. The emergence of variational quantum algorithms (VQAs) provides a practical framework for leveraging today's noisy intermediate-scale quantum (NISQ) processors to solve problems of chemical interest. These hybrid quantum-classical algorithms delegate the computationally demanding task of preparing quantum states to a quantum co-processor, while employing classical optimizers to minimize energy expectation values. This technical guide details the current state of quantum algorithmic approaches, presents quantitative performance benchmarks, and provides detailed experimental protocols for their application in combinatorial chemistry research, framing this progress within the broader thesis of applying VQAs to accelerate drug discovery and materials design.
Table: Key Milestones in Quantum Simulation for Chemistry (2024-2025)
| Date | Institution/Company | Achievement | Significance |
|---|---|---|---|
| March 2025 | IonQ & Ansys | Medical device simulation outperformed classical HPC by 12% [32] | Early documented case of practical quantum advantage in an application |
| April 2025 | Academic Research (arXiv) | Applied VarQITE to the Multiple Knapsack Problem [5] | First use of variational QITE for constrained optimization |
| 2025 | Google Quantum AI | Quantum Echoes algorithm ran 13,000x faster than classical supercomputers [33] | Verifiable quantum advantage with a measurable quantum observable |
| 2025 | Multiple (Industry-wide) | Error rates pushed to record lows of 0.000015% per operation [32] | Critical hardware improvement enabling more complex simulations |
Core Quantum Algorithms for Chemical Simulation
Algorithmic Foundations and Comparative Analysis
At the heart of quantum computational chemistry are algorithms specifically designed to find energy eigenstates and simulate time evolution of molecular systems.
Variational Quantum Eigensolver (VQE): This hybrid algorithm is a cornerstone for near-term quantum chemistry. VQE uses a parameterized quantum circuit (ansatz) to prepare trial wave functions for a molecular system. The quantum computer measures the expectation value of the molecular Hamiltonian, while a classical optimizer adjusts the circuit parameters to minimize this energy, iteratively approximating the ground state [27]. Its resilience to noise makes it particularly suitable for NISQ-era devices.
Quantum Approximate Optimization Algorithm (QAOA): While originally designed for combinatorial optimization, QAOA can be adapted for chemical problems by mapping electronic structure problems to an Ising model [6]. It alternates between applying a cost Hamiltonian (encoding the problem) and a mixer Hamiltonian (exploring the solution space), with parameters classically optimized.
Quantum Imaginary Time Evolution (QITE): A recently advanced alternative, QITE is a ground-state preparation algorithm that has demonstrated improved performance in quantum chemistry problems. Its variational form (VarQITE) has been successfully applied to constrained combinatorial problems, showing significantly lower optimality gaps compared to QAOA [5].
Table: Algorithm Comparison for Chemical Simulation
| Algorithm | Primary Use Case | Key Strength | Near-term Feasibility | Classical Counterpart |
|---|---|---|---|---|
| VQE | Ground-state energy calculation | Noise-resilient, widely demonstrated | High | Full Configuration Interaction |
| QAOA | Combinatorial optimization, mapped chemistry problems | Simple circuit structure | Medium | Simulated Annealing |
| VarQITE | Ground-state preparation, constrained problems | Improved convergence properties | Emerging | Classical Imaginary Time Evolution |
| Quantum Phase Estimation | Precise energy eigenvalue calculation | Exact in theory, high precision | Requires fault-tolerance | Classical Spectral Methods |
Advanced Methodologies: Parallelization and Error Mitigation
As problem sizes approach practical utility, resource limitations necessitate innovative algorithmic strategies. Circuit parallelization has emerged as a critical framework for extending the reach of VQAs. This approach involves splitting a large quantum circuit into smaller, independently executable sub-circuits (slices) that can be run in parallel on limited qubit hardware [6]. The results are then combined classically through a global objective function. For chemical simulations, this enables the treatment of larger molecular active spaces than would fit on a single quantum processor.
Furthermore, error mitigation techniques are essential for extracting meaningful results from current noisy devices. These include readout error mitigation, zero-noise extrapolation, and probabilistic error cancellation. The integration of these techniques with variational algorithms has been crucial for demonstrating chemically accurate results in simulations of small molecules, paving the way for more complex systems [34].
Experimental Protocols and Workflows
Protocol 1: Molecular Energy Calculation Using VQE
This protocol details the steps for calculating the ground state energy of a molecule, a fundamental task in drug discovery for predicting reaction pathways and binding affinities.
Problem Formulation:
- Input: Define the molecular geometry (atomic species and positions) and select a basis set (e.g., 6-311G(d,p)) [35].
- Active Space Selection: Reduce the full molecular Hamiltonian to a manageable size using the Active Space Approximation. A common starting point is a two-electron, two-orbital (2e, 2o) system, which can be represented on a 2-qubit quantum device [35].
- Hamiltonian Generation: Transform the electronic structure problem into a qubit Hamiltonian using a fermion-to-qubit mapping (e.g., Jordan-Wigner or parity transformation).
Quantum Circuit Preparation:
- Ansatz Selection: Choose a parameterized quantum circuit. For initial experiments, a hardware-efficient $R_y$ ansatz with a single layer is often effective [35].
- Parameter Initialization: Set initial parameters, often randomly or based on a classical approximation (e.g., Hartree-Fock state).
Hybrid Execution Loop:
- Quantum Processing: Execute the parameterized circuit on a quantum processor (or simulator). Measure the expectation value of the Hamiltonian terms.
- Classical Optimization: Use a classical optimizer (e.g., COBYLA, SPSA) to adjust the quantum circuit parameters to minimize the energy expectation value.
- Convergence Check: Iterate until energy convergence is achieved within a predefined threshold.
Post-Processing and Validation:
Diagram Title: VQE Hybrid Quantum-Classical Workflow
Protocol 2: Gibbs Free Energy Profile for Prodrug Activation
This protocol outlines a specific application of quantum computing to a critical task in pharmaceutical research: calculating the Gibbs free energy profile for covalent bond cleavage in a prodrug activation process [35].
System Preparation:
- Molecular Geometry Optimization: Perform conformational optimization of the reactant, transition state, and product structures involved in the bond cleavage using classical methods.
- Solvation Model Setup: Implement a solvent model such as the polarizable continuum model (PCM) or the domain decomposition conductor-like screening model (ddCOSMO) to simulate physiological conditions [35].
Quantum Computation of Single-Point Energies:
- For each molecular configuration (reactant, transition state, product), perform a VQE calculation as described in Protocol 1 to obtain the electronic energy within the active space.
Thermochemical Correction:
- Calculate thermal Gibbs corrections (entropic and enthalpic contributions) at the classical level (e.g., HF level) and add them to the quantum-computed electronic energies to obtain the Gibbs free energy [35].
Reaction Profile Construction:
- Plot the Gibbs free energy against the reaction coordinate. The energy barrier is calculated as the difference between the transition state and reactant energies.
Benchmarking:
- Compare the quantum-computed energy barrier with results from Density Functional Theory (DFT) and experimental wet-lab validation to assess predictive accuracy [35].
The Scientist's Toolkit: Essential Research Reagents
This section details the key computational "reagents" and tools required to implement the experimental protocols described above.
Table: Essential Computational Tools for Quantum-Assisted Chemistry
| Tool Category | Specific Example(s) | Function/Purpose | Implementation Note |
|---|---|---|---|
| Quantum Hardware/Simulator | IBM Quantum, Google Quantum AI, SpinQ NMR systems [32] | Provides the physical qubits or high-performance simulation to execute parameterized quantum circuits. | Cloud-based access is standard; educational NMR systems offer hands-on training [32]. |
| Quantum Software Framework | Qiskit, Cirq, TenCirChem [35] | Provides libraries for constructing quantum circuits, performing fermion-to-qubit mapping, and managing hybrid workflows. | TenCirChem was used to implement Gibbs energy workflow with few code lines [35]. |
| Classical Electronic Structure Package | PySCF, Psi4, Gaussian | Handles molecular geometry optimization, basis set generation, and calculation of classical reference energies (HF, CASCI). | Used for pre- and post-processing within the hybrid quantum-classical pipeline. |
| Classical Optimizer | COBYLA, SPSA, L-BFGS-B | The classical component that adjusts quantum circuit parameters to minimize the energy expectation value. | Choice of optimizer impacts convergence speed and resilience to quantum noise. |
| Error Mitigation Toolkit | Readout Error Mitigation, Zero-Noise Extrapolation | Post-processing techniques applied to raw quantum measurements to improve accuracy without physical qubit correction. | Readout error mitigation was applied in the prodrug activation study [35]. |
Performance Benchmarks and Current Capabilities
The field is rapidly progressing from theoretical potential to demonstrable utility. The tables below summarize key quantitative benchmarks that define the current state of the art.
Table: Documented Instances of Quantum Utility in Simulation (2025)
| Application Context | Quantum System Used | Classical Benchmark | Performance Result | Reported By |
|---|---|---|---|---|
| Medical Device Simulation | IonQ 36-qubit computer | Classical HPC | Outperformed by 12% [32] | IonQ & Ansys |
| Out-of-Time-Order Correlator (OTOC) | Google Willow (103 qubits) | Classical Supercomputer | 13,000x faster execution [33] | Google Quantum AI |
| Molecular Geometry via NMR | Google Willow (simulation) | Classical NMR analysis | Improved model of molecular structure [33] | Google & UC Berkeley |
Table: Quantum Hardware Error Correction Milestones (2025)
| Company/Institution | Technology | Key Achievement | Impact on Simulation Fidelity |
|---|---|---|---|
| Willow chip (105 superconducting qubits) | Demonstrated exponential error reduction as qubit count increased ("below threshold") [32] | Enabled beyond-classical complexity algorithms | |
| IBM | Fault-tolerant roadmap | Quantum Starling system (target: 200 logical qubits by 2029) [32] | Path to 100 million error-corrected operations |
| Microsoft | Majorana 1 (topological qubits) | Novel geometric codes showing 1,000-fold error rate reduction [32] | Potential for inherent qubit stability |
| QuEra | Algorithmic fault tolerance | Published techniques reducing error correction overhead by up to 100x [32] | Moves practical quantum computing timelines forward |
The promise of quantum advantage in simulating quantum systems is transitioning from a theoretical certainty to an emerging experimental reality. Variational quantum algorithms serve as the critical bridge, enabling researchers to extract meaningful chemical insights from today's imperfect quantum hardware. The documented instances of utility in calculating molecular energies and simulating reaction profiles for pharmaceutical applications underscore this progress.
The road ahead involves crossing key gaps: from error mitigation to active error correction, from heuristic algorithms to verifiable applications, and from exploratory simulations to credible quantum advantage [34]. As hardware continues to scale—with roadmaps pointing to 1,000+ qubit systems and increasingly sophisticated error correction—the scope of simulatable chemical problems will expand dramatically. For researchers in combinatorial chemistry, engagement with this evolving toolkit is no longer speculative but is becoming a strategic imperative for tackling problems intractable to classical computation. The integration of these quantum simulations into established drug discovery workflows has the potential to significantly accelerate the development of new therapeutics and materials.
From Theory to Practice: Implementing VQAs in Drug Discovery Pipelines
The computation of molecular properties like ground state energy and Gibbs free energy is a central task in chemistry and drug discovery, but conventional classical methods often face significant limitations. These methods, including Density Functional Theory (DFT) and Hartree-Fock (HF), can be computationally demanding for complex systems, struggle to capture quantum effects like entanglement, and can become intractable for large-electron systems [36]. The transformative potential of quantum computing lies in its inherent ability to model quantum mechanical systems more naturally. Quantum algorithms, particularly Variational Quantum Algorithms (VQAs), are designed to overcome these hurdles by offering a more efficient pathway to accurate electronic structure calculations, potentially providing exponential speed-ups over classical methods for certain problems [36].
This technical guide explores the application of VQAs, specifically the Variational Quantum Eigensolver (VQE), for calculating crucial molecular properties. We place this discussion within the broader context of their application to combinatorial chemistry research, where rapidly and accurately evaluating molecular energies can significantly accelerate the screening and design of novel compounds and drugs [37].
Theoretical Foundations: From Molecules to Qubits
The Electronic Structure Problem
The fundamental goal is to solve the electronic Schrödinger equation for a molecule under the Born-Oppenheimer approximation. This involves finding the ground state energy and wavefunction of the molecular Hamiltonian, which describes the system's total energy. Mathematically, the energy of a system is given by the expectation value of the Hamiltonian, ( \hat{H} ): ( E(\theta) = \langle \Psi(\theta) | \hat{H} | \Psi(\theta) \rangle ) [36]. The minimum of this function, ( E{\text{min}} = \min{\theta} E(\theta) ), corresponds to the ground state energy [36].
The Variational Quantum Eigensolver (VQE)
The VQE is a hybrid quantum-classical algorithm that finds the ground state energy of a molecular Hamiltonian. Its operation can be broken down into the following steps [36]:
- Hamiltonian Formulation: The molecular electronic Hamiltonian is written in terms of fermionic creation and annihilation operators.
- Qubit Mapping: This fermionic Hamiltonian is transformed into a qubit Hamiltonian using a fermion-to-qubit transformation (e.g., Jordan-Wigner or parity). The resulting Hamiltonian is a linear combination of Pauli strings: ( \hat{H} = \sumi \mui Pi ), where ( Pi \in {X, Y, Z} ) [36].
- Ansatz Preparation: A parameterized quantum circuit (ansatz), ( U(\theta) ), is used to prepare a trial state on a quantum computer: ( |\Psi(\theta)\rangle = U(\theta)|0\rangle^{\otimes n} ) [36].
- Measurement and Energy Evaluation: The quantum computer measures the expectation values of the individual Pauli terms in the Hamiltonian. These are combined classically to compute the total energy ( E(\theta) ).
- Classical Optimization: A classical optimizer (e.g., Gradient Descent) uses the energy ( E(\theta) ) to update the parameters ( \theta ) iteratively. This loop repeats until convergence, yielding the ground state energy estimate.
Calculating Gibbs Free Energy Profiles
For chemical reactions, such as prodrug activation, calculating the Gibbs free energy profile is critical. It determines the feasibility and rate of reactions, guiding molecular design in drug development [37]. The Gibbs free energy incorporates both potential energy and entropic contributions under specific conditions (e.g., temperature, solvation). In hybrid quantum-classical pipelines, the potential energy surface is computed using a quantum method like VQE, while thermal and solvation corrections are added using classical models [37]. For example, solvation effects can be modeled with a polarizable continuum model (PCM), and thermal Gibbs corrections can be calculated at the HF level [37].
Experimental Protocols and Methodologies
Protocol for Ground State Energy Calculation
The following methodology is adapted from studies comparing VQE to conventional methods like HF and DFT on molecules such as Water (H₂O), Lithium Hydride (LiH), and Methane (CH₄) [36].
- System Selection and Hamiltonian Generation:
- Select the target molecule and its geometry.
- Generate the electronic Hamiltonian in a chosen basis set (e.g., 6-31G, 6-311G(d,p)).
- Active Space Selection and Qubit Mapping:
- Quantum Circuit Configuration:
- Ansatz: Employ a hardware-efficient ansatz, such as an ( N4 ) ( R_y ) ansatz with a single layer, to prepare the trial wave function [37].
- Initial State: Initialize the qubits in the ( |0\rangle^{\otimes n} ) state.
- Execution and Optimization:
- Use the VQE algorithm to measure the energy expectation value.
- Employ a classical optimizer to minimize the energy. The entire workflow can be implemented in quantum chemistry packages like TenCirChem [37].
- Error Mitigation:
- Apply standard techniques like readout error mitigation to improve the accuracy of the quantum measurements [37].
Protocol for Gibbs Free Energy Profile of Prodrug Activation
This protocol is tailored for real-world drug design, exemplified by studying the covalent bond cleavage in a carbon-carbon (C–C) bond-based prodrug activation strategy for β-lapachone [37].
- System Preparation:
- Identify key molecular structures along the reaction coordinate for the C–C bond cleavage.
- Perform conformational optimization of these structures using classical methods.
- Single-Point Energy Calculation:
- For each optimized structure, perform a single-point energy calculation using the VQE protocol outlined in Section 3.1.
- Solvation Treatment:
- Incorporate solvation effects critical for simulating physiological conditions. Use a solvation model like the domain decomposition conductor-like screening model (ddCOSMO) with a water solvent [37].
- Thermal Correction:
- Calculate thermal corrections to Gibbs free energy at the HF level of theory [37].
- Profile Construction:
- Combine the VQE-computed electronic energies, solvation energies, and thermal corrections to construct the final Gibbs free energy profile for the reaction.
Advanced Algorithmic Frameworks
Recent research has introduced novel frameworks to address VQA limitations. The parallel circuit implementation framework splits a large VQA circuit into smaller, independent sub-circuits (slices) that can be trained and executed in parallel. This allows for solving problems larger than the number of available qubits, making more efficient use of near-term hardware [6]. Another innovation is the conditional Generative Quantum Eigensolver (conditional-GQE), which uses a classical encoder-decoder transformer model to generate context-aware quantum circuits. This approach avoids the expressibility limitations of traditional parameterized circuits and can be applied to combinatorial optimization problems derived from chemistry [38].
Results and Data Presentation
Performance Comparison of Electronic Structure Methods
The table below summarizes a comparative analysis of different computational methods for calculating molecular ground state energies, showcasing the performance of VQE against classical techniques [36].
Table 1: Comparison of electronic structure calculation methods for ground state energy.
| Method | Theoretical Foundation | Key Advantages | Key Limitations | Reported Accuracy (for small molecules) |
|---|---|---|---|---|
| Hartree-Fock (HF) | Approximates wavefunction as a single Slater determinant | Computationally efficient, simple model | Neglects electron correlation, often inaccurate | Lower accuracy compared to DFT and VQE [36] |
| Density Functional Theory (DFT) | Uses electron density instead of wavefunction | Good balance of accuracy and speed for many systems | Accuracy depends on functional choice; can struggle with dispersion interactions | Standard accuracy, functional-dependent [36] |
| Variational Quantum Eigensolver (VQE) | Variational principle on a quantum processor | Potentially more accurate modeling of quantum behavior; suited for NISQ devices | Sensitive to ansatz choice, noise, and requires many measurements | Similar or superior accuracy to conventional methods like DFT and HF [36] |
Essential Research Reagent Solutions
The following table details key computational "reagents" and their functions in a quantum computing pipeline for molecular property calculation [37].
Table 2: Research Reagent Solutions for Quantum Computational Chemistry.
| Item / Resource | Function / Purpose in the Workflow |
|---|---|
| Active Space Approximation | Simplifies the quantum chemical problem by focusing computation on a subset of chemically relevant orbitals and electrons, making it feasible for current quantum hardware [37]. |
| Qubit Hamiltonian | The encoded form of the molecular Hamiltonian that can be processed by a quantum device, obtained via fermion-to-qubit transformations (e.g., Jordan-Wigner, parity) [36]. |
| Hardware-Efficient Ansatz | A parameterized quantum circuit designed with native gates of specific quantum hardware, which helps to minimize circuit depth and reduce noise [37]. |
| Classical Optimizer (e.g., GD, SGD) | The classical routine that adjusts the quantum circuit parameters to minimize the energy expectation value, driving the VQE algorithm towards the ground state [36]. |
| Polarizable Continuum Model (PCM) | A classical solvation model used to simulate the effect of a solvent (e.g., water in the human body) on the quantum-mechanical calculation of a molecule [37]. |
| Error Mitigation Techniques | A set of procedures (e.g., readout error mitigation) applied to reduce the impact of noise on results from current imperfect quantum processors [37]. |
Workflow Visualization
VQE for Ground State Energy
The following diagram illustrates the hybrid quantum-classical feedback loop of the Variational Quantum Eigensolver.
Gibbs Free Energy Calculation
This diagram outlines the hybrid workflow for calculating the Gibbs free energy profile of a chemical reaction, such as prodrug activation.
Discussion and Outlook
The application of VQAs for calculating molecular properties represents a significant shift in computational chemistry and drug discovery. The demonstrated ability of VQE to achieve accuracy comparable to conventional methods like DFT and HF, but with potentially fewer resources and a more natural framework for quantum problems, underscores its potential [36]. The successful calculation of Gibbs free energy profiles for real-world drug design problems, such as prodrug activation, further validates the practical utility of hybrid quantum-classical pipelines [37].
Looking forward, overcoming current limitations is key. Challenges include the limited number of qubits, circuit depth restrictions, and the impact of noise. Promising pathways for scaling include circuit parallelization techniques [6] and advanced algorithm frameworks like the conditional-GQE [38]. As quantum hardware continues to advance in scale and fidelity, these algorithms are poised to move from benchmarking small molecules to impacting real-world combinatorial chemistry research, ultimately expediting the development of new materials and therapeutics [36] [37].
The application of variational quantum algorithms (VQAs) in combinatorial chemistry represents a paradigm shift in computational drug discovery [39]. These hybrid quantum-classical algorithms are particularly suited for addressing the complex quantum mechanical problems that underlie molecular interactions, which are computationally prohibitive for classical computers [39] [35]. This technical guide focuses on a specific, real-world application: simulating covalent bond cleavage for prodrug activation, a process critical for targeted cancer therapies.
Prodrug strategies, especially those involving the selective cleavage of robust carbon-carbon (C–C) bonds, offer innovative solutions for improving drug targeting and reducing side effects [35]. Accurately calculating the energy profiles of these reactions is essential for validating their feasibility under physiological conditions. This case study details how a hybrid quantum computing pipeline, centered on the Variational Quantum Eigensolver (VQE), was successfully employed to simulate the Gibbs free energy profile of C–C bond cleavage in a prodrug of β-lapachone, a compound with significant anticancer activity [35].
Computational Foundation: Variational Quantum Algorithms
The Variational Quantum Eigensolver (VQE) Framework
The VQE is a leading hybrid algorithm for the noisy intermediate-scale quantum (NISQ) era [39] [40]. It leverages a classical optimizer to train a parameterized quantum circuit, whose output is used to compute the expectation value of a molecular Hamiltonian [35]. The core principle involves minimizing the energy expectation value to find a variational approximation of the ground state wave function and its energy [35].
For a target molecular system, the electronic Hamiltonian is first mapped to a qubit operator. A parameterized ansatz circuit ( U(\vec{\theta}) ) prepares a trial state ( |\psi(\vec{\theta})\rangle ) from an initial state ( |0\rangle ). The energy expectation value ( E(\vec{\theta}) = \langle \psi(\vec{\theta}) | H | \psi(\vec{\theta}) \rangle ) is measured on the quantum computer. A classical optimizer then adjusts the parameters ( \vec{\theta} ) to minimize ( E(\vec{\theta}) ). Upon convergence, the circuit provides a good approximation of the molecular wave function [35].
Active Space Approximation for Real-World Systems
Simulating large molecules relevant to drug discovery on current quantum hardware requires problem simplification. The active space approximation is a widely used technique that focuses the computational effort on a subset of chemically important electrons and orbitals [35]. In the case of the C–C bond cleavage in β-lapachone prodrug, the active space was reduced to a manageable two-electron/two-orbital system, which can be represented on a 2-qubit quantum device [35]. This approximation allows for the calculation of the CASCI (Complete Active Space Configuration Interaction) energy, considered the exact solution within the defined active space [35].
Case Study: C–C Bond Cleavage in β-Lapachone Prodrug
Background and Chemical Context
The case study investigates an innovative prodrug design for β-lapachone, which utilizes a carbon-carbon bond cleavage mechanism for cancer-specific targeting [35]. This strategy is experimentally validated and aims to enhance the drug's pharmacokinetic and pharmacodynamic properties [35]. The precise calculation of the Gibbs free energy barrier for this bond cleavage is crucial, as it determines whether the reaction will proceed spontaneously at physiological temperatures [35].
Hybrid Quantum Computing Pipeline
The following diagram illustrates the integrated hybrid workflow used in this study, combining classical pre- and post-processing with core quantum computations [35].
Research Reagent Solutions: Computational Tools
The following table details the key computational "reagents" and their functions in the experimental pipeline [35].
| Research Reagent | Function in Experiment |
|---|---|
| Active Space Approximation | Reduces the complex molecular system to a computationally manageable 2-electron/2-orbital model for simulation on near-term quantum devices. |
| 6-311G(d,p) Basis Set | A triple-zeta basis set used in the quantum chemistry calculations to provide a high-quality description of the electronic wavefunction. |
| ddCOSMO Solvation Model | Models the solvation energy within the polarizable continuum model (PCM) framework, simulating the critical effect of a water environment. |
| Hardware-Efficient ( R_y ) Ansatz | A parameterized quantum circuit with a single layer, designed for compatibility with current quantum hardware to prepare the trial state. |
| Readout Error Mitigation | A technique applied during measurement to reduce the impact of inherent hardware noise, enhancing the accuracy of the results. |
Detailed Experimental Protocol
Step 1: System Preparation and Classical Pre-Processing
- Molecular Input: Begin with the molecular structures of the reactant, transition state, and product involved in the C–C bond cleavage, derived from the β-lapachone prodrug system [35].
- Geometry Optimization: Perform a conformational search and optimize the geometry of all species using a classical computational method (e.g., Hartree-Fock) [35].
- Active Space Selection: Define the active space. For this study, a two-electron, two-orbital active space was selected, focusing the quantum computation on the essential orbitals involved in the bond cleavage [35].
- Hamiltonian Generation: Generate the fermionic Hamiltonian for the active space and map it to a qubit Hamiltonian using a parity transformation, making it executable on a quantum processor [35].
Step 2: VQE Execution and Energy Calculation
- Ansatz Initialization: Prepare the trial wave function using a hardware-efficient ansatz. The specific protocol used a single-layer ( R_y ) ansatz, as depicted in the quantum circuit diagram below [35].
- Quantum Execution: Measure the energy expectation value of the qubit Hamiltonian on the quantum device.
- Classical Optimization: Use a classical optimizer (e.g., COBYLA or SPSA) to minimize the energy expectation value by adjusting the ansatz parameters iteratively.
- Convergence Check: Repeat steps 2-3 until the energy converges to a minimum threshold. The resulting energy is the variational ground state energy for the active space [35].
- Solvation and Thermal Correction: Classically compute the solvation energy contribution using the ddCOSMO model and thermal Gibbs corrections at the HF/6-311G(d,p) level of theory. These are added to the VQE energy to obtain the final Gibbs free energy [35].
Results and Benchmarking
The Gibbs free energy profiles for the C–C bond cleavage reaction were successfully computed using the hybrid quantum pipeline. The results were benchmarked against established classical computational methods. The energy barrier computed via VQE was consistent with both classical CASCI results and, crucially, with wet-lab experimental validation, confirming the reaction's feasibility [35]. The following table summarizes a comparative analysis of the computational results.
Table 2: Comparison of Calculated Energy Barriers for C–C Bond Cleavage
| Computational Method | Device/Platform | Energy Barrier (kcal mol⁻¹) | Key Features / Notes |
|---|---|---|---|
| VQE (Quantum) | 2-qubit Superconducting Processor | Consistent with CASCI [35] | Hybrid quantum-classical; used active space approximation and error mitigation. |
| CASCI (Classical) | Classical Computer | Consistent with experiment [35] | Exact solution for the selected active space; benchmark for quantum results. |
| Hartree-Fock (HF) | Classical Computer | Provided reference value [35] | Used for geometry optimization and thermal corrections. |
| Density Functional Theory (DFT) | Classical Computer | Validated in original experimental study [35] | M06-2X functional used in the original study to predict a spontaneously occurring reaction. |
Discussion and Future Directions
Significance in Combinatorial Chemistry
This case study demonstrates a practical application of VQAs to a real-world problem in combinatorial chemistry and drug design [35]. The ability to accurately simulate quantum mechanical processes like covalent bond cleavage directly enables more rational and efficient molecular design. This approach can be extended to screen prodrug candidates in silico, significantly accelerating the early stages of drug development by prioritizing synthesis and testing for the most promising leads [41] [42].
Current Limitations and Challenges
Despite its promise, the field faces several challenges. Current quantum devices are part of the NISQ era, characterized by limited qubit counts, short coherence times, and susceptibility to noise [39] [41]. This restricts the size and complexity of molecules that can be simulated without significant approximation. Furthermore, the scalability of VQEs is hampered by issues like barren plateaus in the optimization landscape and the high measurement overhead required for estimating molecular energies [39] [35].
Future Prospects
The future of quantum computing in drug discovery is closely tied to hardware advancements and algorithmic innovations [32]. Roadmaps from industry leaders project a move towards fault-tolerant quantum computers with hundreds of logical qubits within the next decade [32]. Progress in quantum error correction, as demonstrated by recent breakthroughs in 2025, is rapidly addressing the noise problem [32]. As hardware improves, quantum computing is poised to tackle increasingly complex problems, from modeling full protein-ligand interactions [42] [43] to accelerating the design of novel therapeutics, ultimately reshaping the landscape of pharmaceutical research [41] [32].
The Kirsten rat sarcoma viral oncogene homolog (KRAS) protein is a pivotal signaling molecule, functioning as a GDP-GTP-regulated molecular switch that controls critical cellular processes including proliferation, survival, and differentiation [44]. For decades, KRAS was considered "undruggable" due to its picomolar affinity for GTP/GDP nucleotides and a lack of traditional, targetable binding pockets [45]. This perception has shifted dramatically with the development of covalent inhibitors that specifically target the KRAS G12C mutant, where a glycine-to-cysteine substitution at codon 12 creates a unique vulnerability [44] [45]. The approval of sotorasib (AMG510) and adagrasib (MRTX849) represents a landmark achievement in oncology, validating KRAS as a actionable target, particularly in non-small cell lung cancer (NSCLC) [44].
This case study analyzes the mechanism, efficacy, and resistance profiles of covalent KRAS G12C inhibitors. Furthermore, it explores the emerging role of computational approaches, including variational quantum algorithms (VQAs), in overcoming the combinatorial complexity inherent in multi-parameter drug optimization. These quantum-inspired methods show promise for modeling complex biological systems and optimizing therapeutic strategies against challenging targets like KRAS [6] [38].
KRAS Mutational Landscape and Signaling Biology
Prevalence and Spectrum of KRAS Mutations
KRAS is the most frequently mutated oncogene in human cancers, with mutations found in approximately 30% of all solid tumors [44]. Its prevalence is particularly high in certain malignancies, driven by distinct mutational spectra.
- Pancreatic ductal adenocarcinoma (PDAC): ~82% mutation rate, predominantly G12D (37%) and G12V [44].
- Colorectal cancer (CRC): ~40% mutation rate, with G12D, G12V, and G13D being most common [44].
- Non-small cell lung cancer (NSCLC): ~21% mutation rate, dominated by the G12C subtype (~14% of all NSCLC cases) [44].
These tissue-specific profiles underscore the biological complexity of KRAS-driven oncogenesis and highlight the need for mutation-specific therapeutic strategies.
KRAS Signaling Pathway and Downstream Effectors
KRAS functions as a membrane-bound GTPase, cycling between an active GTP-bound state and an inactive GDP-bound state. This cycle is regulated by guanine nucleotide exchange factors (GEFs, e.g., SOS) which promote GTP loading and activation, and GTPase-activating proteins (GAPs, e.g., NF1) which stimulate GTP hydrolysis to terminate signaling [44]. Oncogenic mutations (e.g., at codons 12, 13, or 61) impair GAP-mediated hydrolysis, trapping KRAS in a perpetually active GTP-bound state [45]. This leads to constitutive activation of downstream effector pathways, primarily the RAF-MEK-ERK (MAPK) pathway and the PI3K-AKT-mTOR axis, which drive uncontrolled cell growth and survival [44] [45].
Diagram: KRAS Signaling Pathway and Downstream Effectors. Oncogenic mutations (e.g., G12C) impair GAP-mediated hydrolysis, locking KRAS in the active state and leading to sustained downstream signaling.
Covalent KRAS G12C Inhibitors: Mechanism and Clinical Profile
Covalent KRAS G12C inhibitors represent a breakthrough in targeted therapy. They exploit the unique cysteine residue present in the G12C mutant to form an irreversible, covalent bond. A critical feature of their mechanism is that they selectively target the GDP-bound, inactive state of KRAS G12C, trapping it in this inactive conformation and preventing it from cycling to the GTP-bound, active state [45].
Table 1: Approved Covalent KRAS G12C Inhibitors and Clinical Efficacy
| Inhibitor (Code Name) | Brand Name | Primary Indication | Objective Response Rate (ORR) | Median Progression-Free Survival (PFS) | Key Resistance Challenges |
|---|---|---|---|---|---|
| Sotorasib (AMG510) | Lumakras | NSCLC (2nd line+) | ~30-40% [44] | ~6 months [44] [45] | Rapid emergence of resistance; limited efficacy in CRC [44] [45] |
| Adagrasib (MRTX849) | Krazati | NSCLC (2nd line+) | ~30-40% [44] | ~6 months [44] [45] | Similar to Sotorasib; on-target secondary mutations, pathway reactivation [45] |
Despite their success, the clinical benefits of these monotherapies are constrained, as evidenced by response rates below 50% and limited median progression-free survival. This underscores the profound challenge of therapeutic resistance [44] [45].
Quantitative Analysis and Resistance Mechanisms
Computational Modeling of Inhibitor Action
Quantitative systems pharmacology (QSP) models have been developed to simulate the complex dynamics of KRAS signaling and inhibitor interactions. These models, formulated as sets of coupled, nonlinear ordinary differential equations, incorporate key parameters to predict steady-state levels of active KRAS (RasGTP) and downstream signaling complexes [46].
Table 2: Key Parameters for Quantitative Systems Pharmacology (QSP) Modeling of KRAS G12C Targeting [46]
| Parameter | Symbol | Value | Units | Description |
|---|---|---|---|---|
| Ras degradation rate | kdeg |
8 × 10⁻⁶ | /s | First-order rate constant for Ras protein turnover (~24h half-life) |
| Ras production rate | kproduction |
3.2 × 10⁻¹² | /Ms | Constant rate of nucleotide-free Ras production |
| On rate for NPI | kon,NPI |
2 × 10⁶ | /Ms | Binding on-rate for Nucleotide Pocket Inhibitors |
| On rate for SIIPI (ARS-853) | kon,SIIPI,ARS |
76 | /Ms | Binding on-rate for Switch-II Pocket Inhibitors (e.g., ARS-853) |
| G12C intrinsic GTPase scaling | ScalingfactorGTPase,G12C |
72 | % | Intrinsic GTP hydrolysis rate of G12C mutant relative to wild-type |
| Cellular GTP abundance | GTP |
1.8 × 10⁻⁴ | M | High intracellular GTP concentration, favoring active KRAS |
These models have provided non-intuitive insights, such as predicting that oncogenic Ras can activate wild-type Ras, a phenomenon later confirmed experimentally [46]. Simulations suggest that optimizing GEF loading could improve the efficacy of nucleotide-competitive inhibitors and that resistance mutations may alter the relative effectiveness of different inhibitor classes [46].
Multidimensional Resistance Mechanisms
Resistance to KRAS G12C inhibitors is not a singular event but a multifaceted adaptive process. The major mechanisms can be categorized as follows:
- On-target mutations: Secondary mutations in KRAS itself (e.g., R68S, Y96C, H95D/Q) that interfere with drug binding while maintaining oncogenicity [45].
- Bypass signaling reactivation: Compensatory activation of parallel or upstream pathways, such as:
- Histologic transformation: Cellular lineage plasticity, such as epithelial-to-mesenchymal transition (EMT) or transformation from adenocarcinoma to squamous cell carcinoma, leading to RAS-independent survival [45].
- Tumor microenvironment (TME) interactions: Immune evasion and stromal support that promote survival despite KRAS inhibition [45].
Diagram: Multidimensional Resistance to KRAS G12C Inhibitors. Resistance arises through on-target alterations, bypass signaling, cellular plasticity, and microenvironment interactions.
The Scientist's Toolkit: Key Reagents and Experimental Methodologies
Table 3: Essential Research Reagents and Resources for KRAS Inhibitor Studies
| Reagent / Resource | Function and Application in KRAS Research |
|---|---|
| KRAS G12C Mutant Cell Lines (e.g., NCI-H358, MIA PaCa-2) | In vitro models for studying inhibitor efficacy, resistance mechanisms, and downstream signaling. Provide a biologically relevant context for testing covalent inhibitor activity [46]. |
| Covalent Inhibitors (Sotorasib, Adagrasib, ARS-853) | Tool compounds for biochemical and cellular assays. ARS-853 is a prototypical Switch-II pocket inhibitor used for mechanistic studies before clinical development [46]. |
| Phospho-Specific Antibodies (p-ERK, p-AKT, p-MEK) | Readouts for downstream pathway activity. Used in Western blotting or immunofluorescence to confirm target engagement and pathway modulation upon KRAS inhibition [45]. |
| QSP Modeling Framework | Computational platform based on ordinary differential equations. Used to simulate Ras activation dynamics, predict inhibitor effects, and test combinatorial strategies in silico before wet-lab validation [46]. |
| Isogenic Paired Cell Lines (Wild-type vs. G12C mutant) | Critical control systems to distinguish on-target effects from off-target toxicity. Generated via CRISPR-Cas9 gene editing [45]. |
Connecting to Variational Quantum Algorithms in Combinatorial Chemistry
The challenge of optimizing combination therapies and predicting multi-parameter resistance landscapes in KRAS-driven cancers represents a problem of immense combinatorial complexity. This is where variational quantum algorithms (VQAs) offer a promising, albeit emerging, conceptual framework [6] [38].
VQAs are hybrid quantum-classical algorithms where a parameterized quantum circuit (ansatz) is optimized by a classical computer to minimize a cost function, which could be the energy of a molecular Hamiltonian [6]. In the context of drug discovery, this approach can be adapted for combinatorial optimization [38]. For example, finding the optimal combination of drugs to overcome a specific resistance profile can be mapped to a Quadratic Unconstrained Binary Optimization (QUBO) problem or an equivalent Ising model, which is the native input for quantum optimization algorithms like the Quantum Approximate Optimization Algorithm (QAOA) [6].
Recent algorithmic advances, such as the conditional Generative Quantum Eigensolver (conditional-GQE), illustrate this bridge. conditional-GQE uses a classical machine-learning model (a transformer) to generate problem-specific quantum circuits that minimize the energy of a given Hamiltonian [38]. When applied to combinatorial optimization, the problem is encoded into an Ising Hamiltonian, and the algorithm generates quantum circuits to find low-energy states corresponding to high-quality solutions [38]. This workflow mirrors the core challenge in KRAS therapy: using a flexible, adaptive algorithm (the generator) to find an optimal configuration (a drug combination) that minimizes a complex objective (tumor viability).
Diagram: Variational Quantum Algorithm for Combinatorial Optimization. This hybrid workflow can be applied to optimize complex systems like multi-drug therapies against resistant KRAS mutants.
The development of covalent KRAS G12C inhibitors has irrevocably changed the landscape of targeted therapy for NSCLC, CRC, and PDAC. However, the limited durability of response due to multifaceted resistance necessitates a new paradigm. The future lies in rational combination therapies informed by a deep understanding of resistance biomarkers and powered by advanced computational models.
The integration of quantitative systems pharmacology and emerging computational techniques, including variational quantum algorithms, provides a powerful framework for tackling the combinatorial complexity of cancer therapy optimization. While practical, large-scale quantum computing in drug discovery is still on the horizon, the conceptual synergy between searching vast chemical and biological spaces and solving complex optimization problems is clear. Continued research into KRAS biology, coupled with advances in these computational fields, will be essential to overcome resistance and achieve lasting remissions for patients with KRAS-driven cancers.
The accurate simulation of molecular electronic structure is a fundamental challenge in quantum chemistry and drug discovery. While the exact solution of the Schrödinger equation provides a complete description, it poses an exponentially scaling problem that becomes computationally intractable for all but the smallest systems [47]. This challenge is particularly acute for studying strongly correlated electrons in complex molecules, where single-reference methods like Hartree-Fock or standard density functional theory (DFT) often fail [48].
The active space approximation has emerged as a powerful strategy to overcome this computational barrier. By strategically focusing computational resources on the most chemically relevant electrons and orbitals, this method enables accurate quantum simulations of molecular systems that would otherwise be prohibitive [47]. With the advent of quantum computing, particularly variational quantum algorithms, the active space approximation provides a crucial bridge between classical computational chemistry and emerging quantum hardware [27].
This technical guide explores the theoretical foundations, implementation methodologies, and practical applications of active space approximation within the context of variational quantum algorithms for combinatorial chemistry research. We present a comprehensive framework that enables researchers to leverage current quantum devices for pharmaceutical development while systematically scaling alongside advancing quantum computing capabilities.
Theoretical Framework
Fundamental Principles of Active Space Approximation
The active space approximation operates on the principle of orbital space separation, dividing the electronic structure problem into fragment and environment degrees of freedom [47]. This approach recognizes that for most chemical processes, only a subset of electrons in specific orbitals actively participate in bond formation, breaking, or electronic excitations.
Mathematically, the electronic Hamiltonian in second quantization is expressed as:
$$\hat{H}=\sum {pq}{h}{pq}{\hat{a}}{p}^{\dagger }{\hat{a}}{q}+\frac{1}{2}\sum {pqrs}{g}{pqrs}{\hat{a}}{p}^{\dagger }{\hat{a}}{r}^{\dagger }{\hat{a}}{s}{\hat{a}}{q}+{\hat{V}}_{nn}\,,$$
where $h{pq}$ and $g{pqrs}$ are one- and two-electron integrals, and ${\hat{a}}{p}^{\dagger }$ and ${\hat{a}}{p}$ are creation and annihilation operators [47].
In active space approximation, this full Hamiltonian is reduced to a fragment Hamiltonian:
$${\hat{H}}^{{\rm{frag}}}=\sum {uv}{V}{uv}^{\text{emb}\,}{\hat{a}}{u}^{\dagger }{\hat{a}}{v}+\frac{1}{2}\sum {uvxy}{g}{uvxy}{\hat{a}}{u}^{\dagger }{\hat{a}}{x}^{\dagger }{\hat{a}}{y}{\hat{a}}{v}\,,$$
where the indices are limited to active orbitals, and the one-electron integrals are replaced by an embedding potential $V_{uv}^{emb}$ that accounts for interactions between inactive and active electrons [47].
Connection to Variational Quantum Algorithms
Variational Quantum Algorithms (VQAs) represent a class of hybrid quantum-classical algorithms designed to leverage current noisy intermediate-scale quantum (NISQ) devices [27]. These algorithms, including the Variational Quantum Eigensolver (VQE) and Quantum Approximate Optimization Algorithm (QAOA), use a quantum computer to prepare and measure parameterized quantum states while employing classical optimization to minimize the energy expectation value [27] [23].
The integration of active space approximation with VQAs creates a powerful synergy: the active space reduces the problem size to fit within quantum hardware constraints, while VQAs provide a means to solve the resulting correlated electron problem with potentially superior efficiency [47]. This combination is particularly valuable for combinatorial chemistry research, where rapid screening of molecular properties is essential for drug discovery [42].
Table 1: Key Variational Quantum Algorithms for Active Space Problems
| Algorithm | Primary Application | Key Features | Compatibility with Active Space |
|---|---|---|---|
| VQE (Variational Quantum Eigensolver) | Ground state energy calculation | Hybrid quantum-classical; resilient to noise | Excellent for CASCI-type problems |
| QAOA (Quantum Approximate Optimization Algorithm) | Combinatorial optimization | Alternating cost and mixer Hamiltonians | Suitable for Ising formulations of active space selection |
| QPE (Quantum Phase Estimation) | Eigenvalue estimation | Requires fault-tolerant quantum computing | Provides high precision for spectral calculations |
| QML (Quantum Machine Learning) | Pattern recognition in chemical space | Enhanced processing of high-dimensional data | Can assist in active space selection |
Methodological Implementation
Quantum Information-Assisted Active Space Selection
Traditional active space selection often relies on chemical intuition, which can be subjective and limited for novel systems. The Quantum Information-Assisted Complete Active Space (QICAS) optimization scheme addresses this challenge by leveraging unique quantum information measures to assess electronic correlation in a predictive manner [48].
The QICAS approach utilizes the von Neumann entropy of orbital reduced density matrices:
$$S(\rhoi) = -\rhoi\log(\rho_i),$$
where $\rhoi = \mathrm{Tr}{\backslash{\phii}}[|\Psi0\rangle\langle\Psi0|]$ is the reduced density matrix for orbital $\phii$, obtained by tracing out all other orbital degrees of freedom from the ground state $|\Psi_0\rangle$ [48]. This entropy quantifies the orbital entanglement and serves as a robust metric for identifying strongly correlated orbitals that should be included in the active space.
The methodological workflow for QICAS implementation involves:
- Initial approximate calculation using affordable multireference methods (e.g., DMRG with low bond dimension)
- Orbital entanglement analysis to identify the most correlated orbitals
- Active space optimization that minimizes the correlation discarded by the approximation
- High-accuracy calculation using the optimized active space
This protocol has demonstrated remarkable efficiency, producing optimized orbital sets for which the CASCI energy reaches the corresponding CASSCF energy within chemical accuracy, and greatly reducing the number of iterations required for CASSCF convergence in challenging systems like the Chromium dimer [48].
Periodic Active Space Embedding for Materials
Many pharmaceutical compounds and catalytic materials exhibit periodic structures that require specialized treatment. The extension of active space embedding to periodic systems through multiconfigurational range-separated DFT (rsDFT) represents a significant advancement [47].
This framework supports both molecular and periodic systems, spin-polarized and unpolarized calculations, and can treat core electrons explicitly or through pseudopotentials [47]. The implementation combines the CP2K package for classical DFT computations with Qiskit Nature for quantum circuit simulations, communicating through message passing interfaces that provide a scalable path toward quantum-centric supercomputing [47].
Active Space Embedding Computational Workflow
Experimental Protocols and Applications
Case Study: Neutral Oxygen Vacancy in Magnesium Oxide
The application of periodic active space embedding coupled with quantum computation has been demonstrated through the accurate prediction of optical properties of the neutral oxygen vacancy in magnesium oxide (MgO) [47]. This system represents a challenging solid-state defect with strong electron correlations.
The experimental protocol for this study involved:
Periodic rsDFT Calculation: A range-separated DFT calculation was performed for the MgO bulk system using the CP2K package, applying the Gaussian and plane waves (GPW) approach [47].
Active Space Definition: The oxygen vacancy defect was treated as the embedded fragment, with an active space consisting of the defect states and nearby ligand orbitals.
Embedded Fragment Hamiltonian Construction: The embedding potential $V_{uv}^{emb}$ was constructed to account for the interactions between the active defect electrons and the inactive environment electrons.
Quantum Circuit Execution: The variational quantum eigensolver (VQE) and quantum equation-of-motion (QEOE) algorithms were deployed through Qiskit Nature to obtain the low-lying spectrum of the embedded fragment Hamiltonian [47].
Spectroscopic Property Calculation: The absorption and emission spectra were computed from the excited states and compared with experimental measurements.
Despite minor discrepancies in the position of the main absorption band, the method demonstrated competitive performance compared to state-of-the-art ab initio approaches, with excellent agreement observed for the experimental photoluminescence emission peak [47].
Table 2: Performance Metrics for Active Space Quantum Simulations
| System | Active Space Size | Algorithm | Accuracy Achieved | Computational Advantage |
|---|---|---|---|---|
| MgO Oxygen Vacancy | Not specified | rsDFT+VQE/QEOE | Excellent agreement with experimental emission peak | Competitive with state-of-the-art ab initio methods |
| Small Correlated Molecules | Variable (QICAS-optimized) | CASCI/CASSCF | Chemical accuracy (≤1 kcal/mol) | CASCI reaches CASSCF energy without orbital optimization |
| Chromium Dimer | Variable (QICAS-optimized) | CASSCF | Greatly reduced iterations | QICAS provides excellent starting point for convergence |
Pharmaceutical Application: KRAS Ligand Discovery
Quantum computing enhanced with active space methods has shown promising applications in drug discovery for challenging targets. A recent study demonstrated the first experimental validation of quantum computing for drug discovery, targeting the KRAS protein, a notoriously difficult cancer drug target [49].
The hybrid quantum-classical workflow implemented:
Classical Machine Learning Pre-screening: A classical computer was used to input a database of molecules experimentally confirmed to bind KRAS, training a machine learning model with this data plus over 100,000 theoretical KRAS binders [49].
Quantum Machine Learning Enhancement: The classical model results were fed into a filter/reward function, and a quantum machine learning model was trained and combined with the classical model to improve generated molecule quality [49].
Iterative Optimization: The researchers cycled between training the classical and quantum models to optimize them in concert [49].
Experimental Validation: The optimized models generated novel ligands predicted to bind KRAS, with two molecules showing real-world potential upon experimental validation [49].
This approach successfully identified ligands for one of the most important cancer drug targets, demonstrating quantum computing's potential to enhance drug discovery by leveraging its inherent ability to simulate quantum mechanical processes more efficiently than classical computers [49].
The Scientist's Toolkit
Implementing active space approximation with variational quantum algorithms requires both theoretical knowledge and practical computational tools. The following table summarizes essential "research reagent solutions" for scientists entering this field.
Table 3: Essential Computational Tools for Active Space Quantum Simulations
| Tool Name | Type | Primary Function | Application in Active Space Methods |
|---|---|---|---|
| CP2K | Classical Software | Ab initio DFT and molecular dynamics | Computes embedding potential for molecular and periodic systems [47] |
| Qiskit Nature | Quantum Software Library | Quantum algorithm implementation for natural sciences | Solves fragment Hamiltonian using VQE and quantum equation-of-motion [47] |
| Quantum Hardware (Various) | Physical Quantum Processor | Execution of quantum circuits | Runs parameterized quantum circuits for active space problems [27] |
| GPT-QE / Conditional-GQE | Classical Generative Model | Quantum circuit generation | Generates context-aware quantum circuits for combinatorial optimization [38] |
| Orbital Entanglement Analysis | Computational Protocol | Active space selection | Identifies strongly correlated orbitals using quantum information measures [48] |
Visualization of Active Space Selection Logic
The logical relationship between different components of an active space calculation can be visualized as a decision workflow, guiding researchers through the key steps of method selection and implementation.
Active Space Selection Logical Workflow
Future Outlook and Strategic Implementation
The integration of active space approximation with variational quantum algorithms represents a rapidly evolving frontier in computational chemistry and drug discovery. As quantum hardware continues to advance, with roadmaps indicating increasingly powerful systems emerging within the next two to five years, these methodologies are expected to transition from proof-of-concept demonstrations to routine tools in pharmaceutical research [42].
For life sciences organizations seeking to harness these capabilities, a strategic approach is essential:
Pinpoint Specific Value Opportunities: Identify pressing R&D challenges where quantum's unique capabilities can create significant benefits, particularly in target discovery and clinical trial efficiency [42].
Build Multidisciplinary Teams: Recruit and cultivate talent with expertise in computational biology, chemistry, and quantum computing to bridge domain knowledge gaps [42].
Establish Strategic Partnerships: Develop networks with quantum technology leaders to access cutting-edge hardware, software, and specialized knowledge [42].
Implement Future-Proof Data Strategies: Establish secure, scalable data infrastructures capable of handling quantum simulation outputs while protecting against emerging quantum decryption threats [42].
The synergy between active space methods and variational quantum algorithms provides a pragmatic pathway for leveraging current NISQ-era quantum devices while systematically scaling toward larger and more complex systems as hardware improves. This approach promises to significantly accelerate drug discovery pipelines, reduce development costs, and ultimately deliver life-changing therapies to patients more rapidly [42] [49].
Hybrid Quantum-Classical Workflows for Quantum Mechanics/Molecular Mechanics (QM/MM) Simulations
Quantum Mechanics/Molecular Mechanics (QM/MM) simulations are a cornerstone of modern computational chemistry, enabling the detailed study of chemical reactions in complex biological environments like proteins and solvents. By treating a critical region of a reaction with quantum mechanics while handling the larger surroundings with molecular mechanics, these methods balance accuracy with computational feasibility [50]. The integration of quantum computing, particularly through Variational Quantum Algorithms (VQAs), is poised to overcome the limitations of classical computational methods. This is especially true for combinatorial chemistry and drug discovery research, where accurately simulating electronic interactions is crucial but often prohibitively expensive for classical computers [35] [51].
This technical guide details the emerging paradigm of hybrid quantum-classical workflows for QM/MM simulations. It is framed within the context of employing VQAs to enhance the accuracy and scope of combinatorial chemistry research, providing researchers and drug development professionals with the methodologies and practical tools needed to implement these cutting-edge approaches.
Core Theoretical Foundations
QM/MM Methodology
The fundamental principle of a QM/MM simulation is the division of the total system into a QM region, where chemical bonds are formed or broken, and an MM region that provides the environmental context [50] [52]. The Hamiltonian for the coupled system in an electrostatic embedding scheme is given by:
[H^{QM/MM} = H^{QM}e -\sumi^n\sumJ^M\frac{e^2QJ}{4\pi\epsilon0r{iJ}} +\sumA^N\sumJ^M\frac{e^2ZAQJ}{4\pi\epsilon0R{AJ}}]
Here, the first term ((H^{QM}_e)) is the Hamiltonian of the isolated QM system. The second and third terms represent the electrostatic interactions between the QM electrons and the MM point charges, and between the QM nuclei and the MM point charges, respectively [50]. This explicit inclusion of MM point charges in the QM Hamiltonian allows the electron density of the QM region to polarize in response to its environment, a critical effect for modeling biological systems.
The Role of Variational Quantum Algorithms
In hybrid workflows, the task of solving the electronic structure problem for the QM region is delegated to a quantum computer. Variational Quantum Algorithms (VQAs) are a class of quantum algorithms designed for the current Noisy Intermediate-Scale Quantum (NISQ) era. They function by using a parameterized quantum circuit (the ansatz) to prepare a trial wavefunction for the molecule [53]. The energy expectation value of this state is measured on the quantum processor, and a classical optimizer is used to adjust the circuit parameters to minimize this energy. Due to the variational principle, the minimized energy approximates the ground state energy of the molecular system [35].
The Variational Quantum Eigensolver (VQE) is the leading VQA for quantum chemistry problems. Its hybrid nature makes it particularly suitable for integration into larger QM/MM workflows, where it can provide the quantum mechanical forces for the QM region [35] [52].
Implementing a Hybrid Quantum-Classical QM/MM Workflow
The following diagram illustrates the integrated pipeline for conducting QM/MM simulations using a variational quantum eigensolver.
System Preparation and Partitioning
The initial step involves preparing the molecular system, such as a protein-ligand complex, and defining the QM and MM regions. The QM region should encompass the part of the system where electronic changes are critical, such as an active site or a covalent bond being formed or broken. For example, in simulating a covalent inhibitor like Sotorasib binding to the KRAS G12C protein, the QM region would include the drug molecule and the cysteine residue involved in the covalent bond [35] [49]. The remaining protein and solvent are treated with an MM force field. Broken bonds between the QM and MM regions are typically capped with hydrogen atoms (link atoms) to satisfy the valence of the QM atoms [50].
Classical Sampling and Configuration Selection
The conformational space of the system is first sampled using classical Molecular Dynamics (MD) simulations with a standard MM force field. This helps capture the flexibility of the protein and solvent. A set of representative molecular configurations is then extracted from this trajectory for subsequent, more accurate QM/MM calculations [51]. This step is crucial because exhaustive sampling with pure QM/MM is computationally prohibitive.
The VQE Quantum Subroutine
For each selected configuration, the electronic energy and forces of the QM region are calculated using the VQE algorithm on a quantum processor. The standard protocol is as follows:
- Active Space Selection: The full electronic structure problem of the QM region is simplified using the active space approximation. A manageable number of electrons and molecular orbitals are selected for the quantum computation (e.g., a 2-electron/2-orbital system) [35].
- Hamiltonian Mapping: The fermionic Hamiltonian of the active space is transformed into a qubit Hamiltonian using a transformation such as Jordan-Wigner or parity.
- Ansatz and Optimization: A parameterized quantum circuit (ansatz), such as the Unitary Coupled Cluster (UCC) or a hardware-efficient ansatz, is prepared. A classical optimizer (e.g., COBYLA, SPSA) iteratively adjusts the parameters to minimize the energy expectation value measured from the quantum device [35] [52].
- Energy and Force Calculation: Once the ground state is approximated, the energy is recorded. The forces on the QM atoms can be calculated using the Hellmann-Feynman theorem or finite-difference methods.
Machine Learning Potential Training
To make large-scale sampling feasible, the sparse QM/MM data generated by the VQE subroutine is used to train a Machine Learning (ML) potential, such as a Neural Network Potential (NNP) [51]. This model learns the relationship between the structure of the QM region in its MM environment and the corresponding QM/MM energy and forces. The trained ML potential can then be used to run efficient MD simulations on the accurate QM/MM potential energy surface, enabling the calculation of thermodynamic properties.
Free Energy Calculation
The final stage uses the ML potential to perform alchemical free energy (AFE) simulations. These methods computationally "transform" one molecule into another (e.g., a ligand from unbound to bound state) through a series of non-physical intermediate states. The resulting free energy difference provides the binding free energy, a key metric for predicting drug efficacy [51].
Quantitative Data and Performance Metrics
The tables below consolidate key quantitative findings and computational parameters from recent pioneering studies.
Table 1: Key Performance Metrics from Recent Hybrid QM/MM Studies
| Study Focus | System Details | QM Method | Key Result / Performance |
|---|---|---|---|
| Prodrug Activation [35] | C-C bond cleavage in β-lapachone prodrug; 2e-/2o active space. | VQE on 2-qubit simulator (with error mitigation). | Successfully computed Gibbs free energy profile for bond cleavage; results consistent with wet-lab validation. |
| Covalent Inhibition [52] | BTK inhibitor binding via Michael addition. | VQE with UCCSD ansatz on GPU simulator (CUDA-Q). | Mapped reaction energy profile for covalent bond formation in a protein active site. |
| Binding Free Energy [51] | MCL-1/19G protein-ligand complex. | ML-potential trained on DFT-based QM/MM data. | Automated workflow for binding free energy prediction for systems requiring quantum treatment. |
| Quantum-Accelerated Screening [49] | KRAS protein ligands. | Hybrid quantum-classical machine learning model. | Identified novel KRAS binders; two experimentally validated. Outperformed classical-only models. |
Table 2: Typical Computational Parameters for VQE in QM/MM
| Parameter | Common Setting / Method | Purpose / Rationale |
|---|---|---|
| Active Space | e.g., (2e, 2o) or (4e, 4o) | Reduces problem size to fit current quantum devices. |
| Qubit Mapping | Parity, Jordan-Wigner | Encodes fermionic Hamiltonian into qubit Hamiltonian. |
| Ansatz | UCCSD, Hardware-Efficient | Parameterized circuit to prepare trial wavefunction. |
| Optimizer | COBYLA, SPSA | Classical routine to minimize energy by varying circuit parameters. |
| Error Mitigation | Readout Error Mitigation | Reduces impact of noise on current quantum hardware. |
The Scientist's Toolkit: Essential Research Reagents & Solutions
The following table details the key software and computational "reagents" required to implement a hybrid QM/MM workflow.
Table 3: Key Software and Computational Tools for Hybrid QM/MM Research
| Tool / Solution | Function / Purpose | Application Context |
|---|---|---|
| GROMACS (with CP2K) [50] | Classical MD engine with interface for QM/MM calculations. | Manages MM dynamics, QM/MM partitioning, and force calculation; interfaces with CP2K for the QM computation. |
| CP2K [50] | Quantum chemistry and solid-state physics software package. | Can act as the QM engine in a classical QM/MM setup; is being extended to interface with quantum computing backends. |
| TenCirChem [35] | Quantum computational chemistry software. | Used for prototyping and running VQE calculations for molecular systems; integrates active space selection and error mitigation. |
| CUDA-Q [52] | A platform for hybrid quantum-classical computing. | Used to simulate and run quantum circuits, particularly VQE, on GPU-based simulators or quantum hardware. |
| Custom ML Potential Codes [51] | Software for training neural network potentials. | Trains models on QM/MM data to create efficient and accurate force fields for free energy calculations (e.g., using eeACSF descriptors). |
Hybrid quantum-classical workflows for QM/MM simulations represent a transformative advancement for computational chemistry and drug discovery. By leveraging the unique capabilities of variational quantum algorithms to solve the electronic structure problem, researchers can achieve unprecedented accuracy in simulating critical processes like covalent drug binding and reaction pathways in physiological environments. While challenges remain in scaling quantum hardware and optimizing algorithms, the methodologies and proof-of-concept results outlined in this guide provide a robust framework for researchers to begin exploring this frontier. The integration of machine learning further bridges the gap between quantum accuracy and the extensive sampling required for reliable free energy estimates, paving the way for these techniques to become standard tools in combinatorial chemistry research.
Overcoming Practical Challenges: Barren Plateaus, Noise, and Circuit Design
Identifying and Mitigating the Barren Plateau Problem in Chemical Ansätze
Variational Quantum Algorithms (VQAs) have emerged as a leading paradigm for leveraging near-term quantum computers to tackle complex problems in quantum chemistry, including the calculation of molecular ground states and electronic properties. [54] [55] These hybrid quantum-classical algorithms function by optimizing the parameters of a parameterized quantum circuit (PQC), or ansatz, to minimize a cost function, often the expectation value of a molecular Hamiltonian. [56] [55]
A formidable challenge to the scalability of VQAs is the barren plateau (BP) phenomenon. In a barren plateau, the gradients of the cost function vanish exponentially with the number of qubits, resulting in a flat optimization landscape where training becomes infeasible. [57] [58] [55] Within the specific context of quantum chemistry, where accurate simulation requires a sufficient number of qubits and circuit depth, the presence of BPs can severely hinder the practical application of VQAs for drug discovery and materials design. [59] This technical guide provides an in-depth analysis of the BP problem specifically for chemical ansätze and details the most current and promising mitigation strategies.
Understanding the Barren Plateau Problem
Fundamental Concepts and Definitions
A Parameterized Quantum Circuit (PQC) is typically expressed as: ( U(\theta) = \prod{i=L}^{1} Wi U(\thetai) ), where ( U(\thetai)=e^{-i\thetai V} ) with a Hermitian operator ( V ), and ( Wi ) are fixed unitary gates. [55] The cost function for a variational quantum eigensolver (VQE), central to quantum chemistry, is the expectation value of the electronic structure Hamiltonian, ( H ): ( C(\theta) = \langle 0 | U^{\dagger}(\theta) H U(\theta) | 0 \rangle ). [56] [55]
A barren plateau is formally characterized by an exponential decay in the variance of the cost function gradient with respect to any parameter ( \thetak ): ( \text{Var}[\partialk C] \in \mathcal{O}(b^{-n}) ), where ( b > 0 ) and ( n ) is the number of qubits. [56] This implies that the probability of determining a non-negligible gradient direction through random sampling becomes exponentially small, stalling the classical optimizer.
Causes of Barren Plateaus in Chemical Ansätze
The BP problem in quantum chemistry applications can originate from several interconnected sources:
- High Expressibility and Unitary 2-Designs: When a PQC is sufficiently expressive and resembles a random unitary circuit (a unitary 2-design), the output states uniformly explore the entire Hilbert space. This randomness leads to a concentration of the cost function value around its mean and the vanishing of gradients. [58] [55]
- Global Cost Functions: The Hamiltonians in quantum chemistry are often global, meaning the observable ( H ) acts non-trivially on all qubits. It has been rigorously proven that global cost functions induce BPs even for shallow-depth circuits. [56]
- Entanglement and Circuit Depth: Highly entangled states, which are common in electronic structure problems, and deep circuit ansätze needed for accurate chemical simulation can exacerbate the BP problem. [55]
- Noise: On current noisy intermediate-scale quantum (NISQ) devices, environmental noise can itself be a source of BPs, further complicating the training process. [56]
Mitigation Strategies for Barren Plateaus
A multi-faceted approach is required to mitigate BPs in chemical applications. The following strategies represent the most current research directions.
Initialization Strategies
Inspired by classical deep learning, intelligent parameter initialization seeks to start the optimization in a region of the landscape with non-vanishing gradients.
- Classical Initialization Heuristics: Recent work has adapted classical initialization schemes like Xavier (Glorot) and He initialization for PQCs. The core idea is to set the initial parameter variance to ( \sigma^2 \propto 1 / n ), where ( n ) is the number of qubits, to help stabilize signal propagation in the quantum circuit. [54]
- Small-Angle Initialization: Initializing parameters to small, random values keeps the initial quantum state close to the identity transformation, preventing the circuit from immediately entering a high-entropy, BP-prone region of the Hilbert space. [54]
- Layerwise and Warm-Start Strategies: Techniques such as layerwise training, where the circuit is grown incrementally, and warm-starting using solutions from classically tractable smaller problems can help avoid BPs by providing a structured starting point. [54] [57]
Table 1: Summary of Initialization Strategies
| Strategy | Core Principle | Key Formulation/Approach | Pros & Cons |
|---|---|---|---|
| Classical Heuristics [54] | Adapt classical NN initialization (Xavier, He) | Set variance ( \sigma_\ell = \sqrt{1/n} ) per circuit layer | Pro: Simple, moderate improvement in some casesCon: Overall benefits can be marginal |
| Small-Angle Init. [54] | Keep circuit near identity | Restrict initial parameters to a narrow range | Pro: Avoids high-entropy statesCon: May limit expressibility |
| Layerwise Training [54] [57] | Incremental circuit growth | Freeze trained layers, add & train new ones | Pro: Retains gradient signalCon: Risk of freezing suboptimal parameters |
Cost Function and Ansatz Design
Modifying the problem itself to be more amenable to training is a powerful approach.
- Local Cost Functions: A highly promising direction is to formulate the problem using local cost functions, where the Hamiltonian is a sum of terms, each acting on at most ( K ) qubits, with ( K ) not scaling with the total number of qubits ( n ). It has been proven that for shallow circuits ( L = \mathcal{O}(\log n) ), local cost functions do not exhibit barren plateaus: ( \text{Var}[\partial_k C] = \Omega(1/\text{poly}(n)) ). [56]
- Symmetry-Preserving Ansätze: Designing ansätze that inherently preserve the physical symmetries of the molecular Hamiltonian (e.g., particle number, spin symmetry) restricts the optimization to a relevant subspace of the full Hilbert space, which can prevent gradient vanishing. [54] [57]
- Problem-Informed and Shallow Ansätze: Using ansätze that incorporate domain knowledge of quantum chemistry (e.g., the Unitary Coupled Cluster ansatz) and striving for shallower circuit architectures can reduce the propensity for BPs. [54] [57]
Non-Unitary and Engineered Dissipation
A radical departure from purely unitary evolution involves the strategic introduction of non-unitary operations.
- Engineered Dissipation: This method adds a properly engineered Markovian dissipation channel, modeled by a GKLS (Gorini-Kossakowski-Sudarshan-Lindblad) master equation, after each unitary layer of the variational circuit. The dissipative map, ( \mathcal{E}(\sigma) = e^{\mathcal{L}(\sigma) \Delta t} ), is designed to effectively transform the global cost function problem into a local one, thereby mitigating the BP. [56]
- Non-Unitary Ansatz: The overall circuit becomes a sequence of unitary and non-unitary steps: ( \Phi(\sigma, \theta)\rho = \mathcal{E}(\sigma)[U(\theta)\rho U^{\dagger}(\theta)] ). The parameters of both the unitary gates ( U(\theta) ) and the dissipator ( \mathcal{L}(\sigma) ) are optimized. This approach has shown promise in mitigating BPs for quantum chemistry problems. [56]
Algorithmic and Structural Innovations
Other innovative strategies are emerging from the broader VQA research community.
- Generative Quantum Eigensolvers (GQE): This approach circumvents the parameterized circuit paradigm altogether. A classical generative model (e.g., a transformer) is trained to generate sequences of quantum gates that form a circuit. The parameters reside in the classical model, not the quantum circuit, offering a novel way to escape expressibility limitations of traditional PQCs. [38]
- Circuit Splitting and Parallelization: For large problems, the quantum circuit can be split into smaller, independent "slices" that are executed and optimized in parallel on a quantum processor with limited qubits. A global objective function then guides the optimization of these parallel slices. [6]
Table 2: Overview of Key Mitigation Strategies Beyond Initialization
| Strategy | Core Principle | Key Formulation/Approach | Theoretical Guarantee |
|---|---|---|---|
| Local Cost Functions [56] | Use Hamiltonians with local terms | ( H = \sumi ci Hi ), where ( Hi ) acts on K qubits | For ( L=\mathcal{O}(\log n) ), ( \text{Var}[\partial_k C] = \Omega(1/\text{poly}(n)) ) |
| Engineered Dissipation [56] | Use non-unitary maps to create local effective cost | ( \Phi(\sigma, \theta)\rho = e^{\mathcal{L}(\sigma)\Delta t}[U(\theta)\rho U^\dagger(\theta)] ) | Mitigates BPs by breaking unitary 2-design assumption |
| Generative Quantum Eigensolver (GQE) [38] | Classical AI generates circuit structures | Transformer decoder outputs gate sequences; trained via Direct Preference Optimization (DPO) | Avoids expressibility limits and embedding issues of PQCs |
Experimental Protocols and Validation
Protocol: Mitigating BPs via Engineered Dissipation
This protocol outlines the steps for implementing the engineered dissipation strategy for a chemical Hamiltonian, based on the work of [56].
- Problem Formulation: Define the target molecular Hamiltonian ( H ) (global) and choose a parameterized unitary ansatz ( U(\theta) ).
- Dissipator Design: Design a parameterized dissipator ( \mathcal{L}(\sigma) ). The required form is a sum of local jump operators ( Aj ) such that the steady state of ( \mathcal{L} ) is a state for which ( H ) acts as a local operator. A common choice is ( \mathcal{L}(\sigma)\rho = \sumj \gammaj (Aj \rho Aj^\dagger - \frac{1}{2}{Aj^\dagger Aj, \rho}) ), where parameters ( \sigma ) include the rates ( \gammaj ).
- Circuit Implementation: Construct the non-unitary ansatz ( \Phi(\sigma, \theta)\rho = e^{\mathcal{L}(\sigma)\Delta t}[U(\theta)\rho U^{\dagger}(\theta)] ). On near-term hardware, this may be implemented using probabilistic techniques or by coupling to ancillary qubits that are subsequently measured and reset.
- Optimization Loop: a. Prepare the initial state ( \rho{in} ). b. Apply the non-unitary ansatz ( \Phi(\sigma, \theta) ). c. Measure the expectation value ( C(\theta, \sigma) = \text{Tr}[H \Phi(\sigma, \theta)\rho{in}] ). d. Use a classical optimizer (e.g., gradient-based) to update both parameter sets ( \theta ) and ( \sigma ) to minimize ( C ).
- Validation: Compare the convergence speed and the final energy error against a purely unitary VQE baseline. A successful mitigation will show a slower decay of gradient variance as the number of qubits increases and a more reliable convergence to the ground state.
Protocol: Applying Local Cost Functions
This protocol describes how to leverage local cost functions to avoid BPs, applicable when a local formulation of the problem is feasible. [56]
- Hamiltonian Decomposition: Decompose the global molecular Hamiltonian ( H ) into a sum of ( K )-local terms: ( H = c0 I + \sum{i=1}^N ci Hi ), where each ( H_i ) acts non-trivially on at most ( K ) qubits.
- Cost Function Definition: Instead of a single global cost ( Cg(\theta) = \text{Tr}[H U(\theta)\rho{in} U^\dagger(\theta)] ), define a local cost function as a weighted sum of local expectations: ( Cl(\theta) = \sumi ci \cdot \text{Tr}[Hi U(\theta)\rho_{in} U^\dagger(\theta)] ).
- Circuit and Measurement: a. Use a shallow-depth ansatz where the number of layers ( L = \mathcal{O}(\log n) ). b. For each iteration of the optimization loop, measure the expectation values of the local observables ( H_i ) (or a subset thereof). This can be done more efficiently than measuring the full global Hamiltonian.
- Optimization: Update the parameters ( \theta ) by computing the gradient of the local cost function ( C_l(\theta) ). The variance of these gradients is guaranteed to decay at worst polynomially with ( n ), preventing BPs.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential "Reagents" for BP Mitigation Experiments
| Tool / Technique | Function / Purpose | Example in Chemical Context |
|---|---|---|
| Local Cost Function [56] | Prevents BP by restricting cost to local observables | Decompose molecular Hamiltonian ( H ) into a sum of few-qubit Pauli strings |
| Parameter Shift Rule [58] | Enables exact gradient calculation on quantum hardware | Compute ( \partial C/\partial\theta_i ) for UCC-type ansätze without finite-difference approximations |
| Graph Neural Network (GNN) Encoder [38] | Encodes molecular structure into generative model | In Conditional-GQE, a GNN processes molecular graph to condition circuit generation |
| Direct Preference Optimization (DPO) [38] | Trains generative models without labeled datasets | Optimizes a classical transformer to generate high-performing quantum circuits for a molecule |
| Hardware-Efficient Ansatz | Reduces circuit depth for NISQ devices | A layered ansatz of single-qubit rotations and entangling gates native to the target quantum processor |
| Layerwise Learning [57] | Mitigates BPs via incremental training | Train a minimal ansatz for a small active space of a molecule, then progressively add layers and re-optimize |
The barren plateau problem represents a significant obstacle to applying VQAs to realistic quantum chemistry problems. No single solution is a panacea; however, a rich and evolving toolkit of strategies provides a clear path forward. Promising directions include the formulation of local cost functions, the strategic use of engineered dissipation to break the unitary 2-design assumption, and innovative generative models that bypass the limitations of traditional parameterized circuits. For researchers in drug development and combinatorial chemistry, a pragmatic approach that combines intelligent parameter initialization with problem-informed, shallow ansätze is a viable starting point. As quantum hardware and algorithms co-evolve, the systematic mitigation of barren plateaus will be instrumental in unlocking the full potential of quantum computing for transformative chemical research.
In the pursuit of quantum advantage for combinatorial chemistry research, Variational Quantum Algorithms (VQAs) have emerged as the most promising framework for noisy intermediate-scale quantum (NISQ) devices. These hybrid quantum-classical algorithms leverage parameterized quantum circuits, known as ansätze, to approximate solutions to computationally hard problems. The fundamental variational principle states that for any trial wavefunction |ψ(θ)⟩ and a Hamiltonian H, the expectation value ⟨ψ(θ)|H|ψ(θ)⟩ provides an upper bound to the ground state energy E_g, forming the theoretical foundation for VQAs [60] [61]. In chemical research, this enables the estimation of molecular properties and reaction pathways that are classically intractable.
The selection of an appropriate ansatz represents one of the most critical design decisions researchers face, balancing expressibility, trainability, and hardware efficiency. This technical guide examines the two predominant strategies: problem-inspired circuits that incorporate domain knowledge of the chemical system, and hardware-efficient designs that maximize fidelity on imperfect quantum devices. We provide a structured framework for ansatz selection, supported by quantitative performance data and detailed experimental protocols tailored to drug development professionals seeking to leverage quantum computational chemistry.
Ansatz Selection Framework: A Comparative Analysis
Fundamental Taxonomy and Characteristics
The ansatz landscape for quantum computational chemistry divides broadly into two philosophical approaches with distinct advantages and limitations:
Problem-Inspired Ansätze incorporate specific knowledge of the target problem directly into their structure. For quantum chemistry applications, this typically involves designing circuits based on the molecular Hamiltonian itself. Examples include the Unitary Coupled Cluster (UCC) ansatz, which encodes excitations from a reference state, and the Hamiltonian Variational Ansatz (HVA), which leverages the problem Hamiltonian to construct evolution pathways [62] [61]. These approaches maintain strong physical relevance but often require deeper circuits that challenge current NISQ device capabilities.
Hardware-Efficient Ansätze prioritize device compatibility by employing native gate sets and connectivity patterns specific to the target quantum processor. Rather than mirroring problem structure, these circuits emphasize expressibility within hardware constraints, typically featuring alternating layers of single-qubit rotations and entangling gates that respect the processor's topology [61]. While more resilient to noise, they face challenges with barren plateaus where gradients vanish exponentially with system size [62] [61].
Table 1: Core Characteristics of Ansatz Paradigms
| Feature | Problem-Inspired Ansätze | Hardware-Efficient Ansätze |
|---|---|---|
| Design Principle | Incorporates domain knowledge of Hamiltonian | Optimized for specific quantum hardware |
| Circuit Depth | Typically higher [62] | Shallower, more NISQ-friendly [63] |
| Parametric Efficiency | Higher (physically motivated parameters) | Lower (may require more parameters) |
| Trainability | Prone to barren plateaus for complex systems [61] | Highly susceptible to barren plateaus [62] [61] |
| Hardware Requirements | Demanding of coherence times and gate fidelities | Tolerant of moderate noise levels |
| Interpretability | High (parameters map to physical quantities) | Lower (parameters lack physical meaning) |
Performance Metrics and Benchmarking
Recent experimental studies provide quantitative insights into ansatz performance across various chemical and optimization problems. The approximation ratio - defined as the ratio between the solution quality obtained and the theoretical optimum - serves as a key metric for evaluating effectiveness [63].
In Max-Cut optimization benchmarks, problem-inspired approaches like the Quantum Approximate Optimization Algorithm (QAOA) have demonstrated approximation ratios of up to 0.78 for 100-vertex problems on actual hardware [63]. Meanwhile, novel hardware-efficient approaches like the Linear Chain QAOA (LC-QAOA) have achieved significant performance improvements by restructuring entangling gates along a linear path to minimize circuit depth and execution time [63]. The Heat Exchange (HE) ansatz, inspired by algorithmic cooling principles, has demonstrated superior approximation ratios compared to conventional hardware-efficient and QAOA ansatzes for complete network Max-Cut problems while maintaining hardware compatibility [62].
Table 2: Experimental Performance Comparison of Recent Ansatz Implementations
| Ansatz Type | Problem | Performance Metric | Qubit Count |
|---|---|---|---|
| Linear Chain QAOA [63] | Max-Cut on random regular graphs | 0.78 approximation ratio | 100 |
| Heat Exchange Ansatz [62] | Complete network Max-Cut | Superior approximation ratio vs. hardware-efficient/QAOA | N/S |
| iHVA with QITE [64] | Multiple Knapsack Problem | Significantly lower optimality gap vs. QAOA | N/S |
| Conditional-GQE [38] | Combinatorial optimization | ~99% accuracy on 10-qubit problems | 10 |
Problem-Inspired Ansätze: Domain-Knowledge Integration
Quantum Alternating Operator Ansatz (QAOA) for Combinatorial Optimization
The Quantum Approximate Optimization Algorithm and its generalization, the Quantum Alternating Operator Ansatz (QAOA+), provide a structured framework for mapping combinatorial chemistry problems to quantum circuits [64]. The algorithm employs alternating operators derived from the problem Hamiltonian and a mixing Hamiltonian:
|ψp(γ,β)⟩ = e^{-iβpHM}e^{-iγpHC}⋯e^{-iβ1HM}e^{-iγ1H_C}|+⟩^{⊗N}
Where HC is the cost Hamiltonian encoding the optimization problem, HM is the mixing Hamiltonian, and (γ,β) are variational parameters optimized classically [63]. For molecular energy calculations, the cost Hamiltonian is derived from the electronic structure problem through transformations such as the Jordan-Wigner or Bravyi-Kitaev mapping [61].
A recent innovation, the Linear Chain QAOA (LC-QAOA), modifies this approach by identifying a long path within the problem graph and establishing entanglement only between adjacent qubits along this path. This strategy creates a hardware-native linear topology that eliminates SWAP overhead, enabling depth-independent circuit execution times that scale favorably for large problems [63]. In experimental implementations, this approach achieved an approximation ratio of 0.78 on non-hardware-native random regular MaxCut instances with 100 vertices using 100 qubits, demonstrating its potential for scalable quantum optimization [63].
Unitary Coupled Cluster (UCC) and Hamiltonian Variational Ansatz (HVA)
For quantum chemistry applications, the Unitary Coupled Cluster ansatz provides a chemically motivated approach that systematically improves upon a reference state (typically Hartree-Fock) by including excitation operators:
U(θ) = exp(∑θi(Ti - T_i^†))
where Ti represents excitation operators and θi are variational parameters [61]. While highly accurate, UCC circuits can become deep and challenging to implement on current hardware, prompting investigations into truncated versions and approximations.
The Hamiltonian Variational Ansatz (HVA) structures the quantum circuit based on the problem Hamiltonian itself, offering a balance between physical relevance and implementability. Recent studies have shown that problem-inspired ansatzes like HVA can mitigate barren plateau issues compared to highly expressive hardware-efficient circuits, as their structured nature constrains the optimization landscape to physically relevant regions [62].
Hardware-Efficient Ansätze: Practical Implementation on NISQ Devices
Topology-Aware Circuit Design
Hardware-efficient ansatzes explicitly account for the physical constraints of quantum processors, including qubit connectivity, native gate sets, and coherence limitations. The fundamental design principle involves creating parameterized circuits using primarily gates native to the target hardware, minimizing the need for costly decompositions or SWAP networks [61].
The Linear Chain QAOA exemplifies this approach by restructuring the quantum circuit to match hardware topology. By identifying a linear chain within the problem graph and placing entangling gates sequentially along this chain, the ansatz achieves shallow quantum circuits with execution times that scale independently of problem size [63]. This strategy has demonstrated particular value for large-scale problems where SWAP overhead would otherwise dominate circuit depth.
The Heat Exchange (HE) ansatz represents another innovative hardware-efficient approach, drawing inspiration from algorithmic cooling principles. This minimalistic ansatz facilitates efficient population redistribution without requiring bath resets, simplifying implementation on NISQ devices while achieving superior approximation ratios for optimization problems compared to conventional hardware-efficient and QAOA ansatzes [62].
Error Resilience and Trainability Considerations
While hardware-efficient ansatzes offer practical advantages for NISQ implementation, they face significant challenges with barren plateaus - regions in the optimization landscape where gradients vanish exponentially with system size [61]. The high expressibility of these circuits, while beneficial for exploring the solution space, contributes to this phenomenon and can hinder practical trainability for large problems.
Recent research has explored various strategies to mitigate these issues:
- Layer-wise training approaches that optimize parameters sequentially rather than simultaneously
- Gradient-based optimization methods specifically designed for quantum landscapes [61]
- Problem-informed initialization that leverages classical approximations to start near promising regions of the parameter space
- Noise-aware compilation techniques that account for device-specific error profiles [63]
Experimental Protocols and Research Reagent Solutions
Implementation Methodology for Chemistry Applications
Successfully implementing variational quantum algorithms for combinatorial chemistry requires careful attention to both algorithmic design and experimental practicalities. The following protocol outlines a standardized approach for ground state energy calculation of molecular systems:
Problem Formulation: Transform the molecular electronic structure problem into a qubit Hamiltonian using second quantization and fermion-to-qubit mapping (Jordan-Wigner or Bravyi-Kitaev) [61].
Ansatz Selection: Choose an appropriate ansatz based on molecular size, available hardware, and precision requirements. For small molecules (≤ 10 qubits), UCCSD offers high accuracy; for larger systems, hardware-efficient variants or QAOA-derived approaches provide more practical alternatives.
Parameter Initialization: Initialize parameters using classical approximations (Hartree-Fock, coupled cluster) for problem-inspired ansatzes or strategic initialization (e.g., near identity operations) for hardware-efficient approaches to avoid barren plateaus.
Quantum Execution: Execute parameterized circuits on quantum hardware or simulator, measuring expectation values of the Hamiltonian terms. Employ measurement reduction techniques (qubit tapering, grouping) to minimize resource requirements [61].
Classical Optimization: Employ classical optimizers (SPSA, COBYLA, BFGS) to minimize energy expectation value. For hardware-efficient ansatzes, consider layer-wise training or progressive growing strategies to enhance convergence [61].
Error Mitigation: Apply readout error mitigation, zero-noise extrapolation, or probabilistic error cancellation to enhance result quality without quantum error correction [61].
Research Reagent Solutions: Essential Components for Quantum Computational Chemistry
Table 3: Essential Research Components for VQA Implementation in Chemistry
| Component | Function | Implementation Examples |
|---|---|---|
| Molecular-to-Qubit Mapping | Encodes electronic structure problem into qubit Hamiltonian | Jordan-Wigner, Bravyi-Kitaev transformations [61] |
| Variational Circuit Templates | Parameterized quantum circuits for ansatz exploration | UCCSD, HVA, Hardware-Efficient Ansatz (HEA) [62] [61] |
| Classical Optimizers | Adjusts circuit parameters to minimize energy | SPSA, COBYLA, BFGS, quantum natural gradient [61] |
| Error Mitigation Techniques | Counteracts hardware noise without full error correction | Readout calibration, zero-noise extrapolation, probabilistic error cancellation [61] |
| Quantum Simulators | Validates algorithms before hardware deployment | Qiskit, Cirq, Pennylane with statevector/density matrix simulations [61] |
| Measurement Reduction Tools | Minimizes number of measurements required | Hamiltonian term grouping, qubit tapering, classical shadows [61] |
Emerging Approaches and Future Directions
Algorithmic Innovations and Hybrid Strategies
The evolving landscape of ansatz design reflects a growing convergence between problem-inspired and hardware-efficient approaches. Several promising directions are emerging:
QITE-Inspired Ansatzes: Quantum Imaginary Time Evolution (QITE) provides an alternative foundation for ansatz design, with variational implementations (VarQITE) demonstrating improved performance for constrained optimization problems [64]. The imaginary Hamiltonian Variational Ansatz (iHVA) adapts this approach specifically for optimization problems, achieving significantly lower optimality gaps compared to QAOA on problems like the Multiple Knapsack Problem [64].
Conditional Generative Models: The Conditional Generative Quantum Eigensolver (conditional-GQE) represents a paradigm shift in ansatz design, employing a classical generative model (encoder-decoder transformer) to generate context-aware quantum circuits [38]. This approach achieves approximately 99% accuracy on 10-qubit combinatorial optimization problems and demonstrates the potential for leveraging classical machine learning to design problem-specific quantum circuits [38].
Adaptive Ansatz Construction: Algorithms like ADAPT-VQE and ADAPT-QAOA dynamically construct circuits by selecting operators from a pool based on gradient information, potentially offering improved efficiency by eliminating redundant parameterized gates [61].
Co-Design for Next-Generation Quantum Chemistry
As quantum hardware continues to evolve, with processors like IBM's Heron supporting up to 5,000 gate operations [62], the strategy of hardware-algorithm co-design becomes increasingly critical. Future ansatz development will likely focus on:
- Hardware-Specific Compilers that optimize circuit depth and fidelity for particular processor architectures
- Noise-Adaptive Circuits that explicitly account for and mitigate specific error profiles of target devices
- Problem-Tailored Entanglement Strategies that balance expressibility and trainability for specific chemical applications
- Classical-Quantum Hybrid Representations that leverage classical machine learning to enhance quantum circuit effectiveness [38]
The integration of these approaches promises to extend the reach of variational quantum algorithms to increasingly complex chemical systems, potentially accelerating drug discovery and materials design in the coming years.
Optimization Techniques for Parameterized Quantum Circuits
Parameterized Quantum Circuits (PQCs) represent a foundational component of variational quantum algorithms (VQAs), which have emerged as the most promising approach for leveraging current noisy intermediate-scale quantum (NISQ) devices. Within combinatorial chemistry research, PQCs enable the approximation of molecular ground states and properties through a hybrid quantum-classical framework, offering a potential pathway to quantum advantage for problems that remain intractable for classical computers. The optimization of PQC parameters is therefore not merely a technical consideration but a central determinant of algorithmic success, particularly for complex chemical systems like the Heisenberg model, Fermi-Hubbard model, and drug-target interactions where accurate simulation is critical. This guide provides a comprehensive technical examination of contemporary PQC optimization techniques, with a specific focus on methodologies demonstrating efficacy for chemical applications.
Fundamental Concepts of Parameterized Quantum Circuits
Mathematical and Structural Formalism
A Parameterized Quantum Circuit is formally defined as a unitary transformation, ( U(\boldsymbol{\theta}) ), composed of a sequence of parameterized gate operations [65]. The typical structure consists of alternating layers of parameterized single-qubit gates and fixed entangling gates, often arranged in a repeating pattern of ( L ) layers. This structure can be expressed as: [ U(\boldsymbol{\theta}) = U{L-1}(\boldsymbol{\theta}{L-1}) \cdots U1(\boldsymbol{\theta}1) U0(\boldsymbol{\theta}0) ] where each layer ( Ul(\boldsymbol{\theta}l) ) is further decomposable into an entangling block, ( Wl ), and a product of single-qubit rotations [65]: [ Ul(\boldsymbol{\theta}l) = Wl \left( \bigotimes{k=1}^{n} e^{-i\theta{ln+k} H{ln+k}/2 \right) ] In this formulation, ( n ) denotes the number of qubits, ( H{ln+k} ) is a Hermitian operator acting on the ( k )-th qubit in the ( l )-th layer, and ( \theta{ln+k} ) is the continuous parameter optimized by a classical algorithm. The entangling layer ( Wl ), frequently composed of CNOT or controlled-Z gates, generates the quantum correlations essential for capturing non-trivial quantum states relevant to molecular systems.
The Role of PQCs in Variational Quantum Algorithms
In the VQA paradigm for combinatorial chemistry, a PQC prepares a trial wavefunction ( |\psi(\boldsymbol{\theta})\rangle = U(\boldsymbol{\theta}) |0\rangle^{\otimes n} ) that approximates the ground state of a target molecular Hamiltonian, ( H ), derived from the electronic structure problem [35]. The core variational principle involves minimizing the energy expectation value: [ E(\boldsymbol{\theta}) = \langle \psi(\boldsymbol{\theta})| H |\psi(\boldsymbol{\theta})\rangle ] This minimization is performed by a classical optimizer that iteratively adjusts the parameters ( \boldsymbol{\theta} ) based on measurements from the quantum computer. The quality of this approximation directly impacts the accuracy of predicted molecular properties, reaction pathways, and drug-binding affinities.
Classical Optimization Methods for PQCs
The optimization landscape of PQCs is characterized by high dimensionality, noise, and the presence of barren plateaus, making the choice of classical optimizer critical. The following table summarizes the primary optimization families and their characteristics.
Table 1: Classification of Classical Optimizers for Parameterized Quantum Circuits
| Optimizer Class | Representative Algorithms | Key Mechanism | Advantages | Limitations |
|---|---|---|---|---|
| Gradient-Based | Adam, Stochastic Gradient Descent, Quantum Natural Gradient [65] | Computes gradients via parameter-shift rule [65] | Fast convergence in smooth landscapes; widely used | Prone to converge to local minima; requires many circuit evaluations |
| Sequential Coordinate Descent | Rotosolve, Rotosolve-based methods [65] [66] | Optimizes each parameter sequentially while keeping others fixed [66] | Reduced circuit evaluations per step; avoids gradient estimation | May require more optimization iterations overall |
| Sparsity-Inducing | Rotolasso [66] | Coordinate descent with L1-regularization to drive parameters to zero [66] | Reduces active parameter count; simplifies circuits | Requires tuning of regularization hyperparameter |
| Hybrid Adaptive | Early-Stopping Hybrids [65] [67] | Dynamically switches optimizers based on cost function progress [65] | Robustness; balances exploration and exploitation | Increased algorithmic complexity |
Detailed Methodologies and Protocols
Gradient-Based Optimization with Parameter-Shift Rule
The parameter-shift rule is a cornerstone technique for evaluating exact gradients of PQCs on quantum hardware [65]. For a parameter ( \thetai ) in a gate ( e^{-i\thetai Hi/2} ), the gradient of the cost function ( C(\boldsymbol{\theta}) ) is given by: [ \nablai C(\boldsymbol{\theta}) = \frac{C(\boldsymbol{\theta} + \frac{\pi}{2}\boldsymbol{e}i) - C(\boldsymbol{\theta} - \frac{\pi}{2}\boldsymbol{e}i)}{2} ] where ( \boldsymbol{e}_i ) is the unit vector in the ( i )-th direction. This protocol enables the use of powerful classical optimizers like Adam [65]. The standard experimental procedure is as follows:
- Initialization: Prepare an initial parameter vector ( \boldsymbol{\theta}^{(0)} ), often randomly.
- Gradient Evaluation: For each parameter ( \thetai ), execute the circuit at ( \boldsymbol{\theta}^{(t)} \pm \frac{\pi}{2}\boldsymbol{e}i ) to estimate the two expectation values required by the parameter-shift rule. This typically requires ( 2p ) circuit executions per iteration, where ( p ) is the number of parameters.
- Parameter Update: Apply the classical optimizer (e.g., Adam) to compute the new parameter vector ( \boldsymbol{\theta}^{(t+1)} ) using the estimated gradient.
- Iteration: Repeat steps 2-3 until convergence of the cost function ( C(\boldsymbol{\theta}) ) or until a maximum number of iterations is reached.
Sparsity-Inducing Coordinate Descent (Rotolasso)
Rotolasso integrates coordinate descent with an L1-regularization term to promote sparsity in the parameter vector [66]. The cost function is modified as: [ C{\text{reg}}(\boldsymbol{\theta}) = C(\boldsymbol{\theta}) + \lambda \|\boldsymbol{\theta}\|1 ] where ( \lambda ) is the regularization strength. The protocol proceeds as follows:
- Cyclic Parameter Selection: Iterate through parameters ( \theta1, \theta2, \ldots, \theta_p ) in a fixed order.
- Univariate Optimization: For the current parameter ( \thetai ), minimize the cost function ( C{\text{reg}}(\boldsymbol{\theta}) ) with respect to ( \thetai ) only, using a direct search method like Rotosolve. This involves evaluating the cost at multiple values of ( \thetai ) to find the optimum.
- Thresholding: Apply a hard threshold to set ( \theta_i = 0 ) if its optimized value falls below a predefined small epsilon ( \epsilon ), effectively removing the corresponding gate from the active circuit.
- Iteration and Convergence: Cycle through all parameters until the change in the regularized cost function ( C_{\text{reg}}(\boldsymbol{\theta}) ) is negligible.
This method significantly reduces the number of circuit evaluations per iteration compared to gradient-based methods and yields simpler, more NISQ-friendly circuits [66].
Advanced Hybrid and Adaptive Methods
Early Stopping Hybrid Algorithms
Recent research has introduced hybrid optimizers that dynamically switch between a less expressive but computationally efficient optimizer (e.g., Rotosolve) and a more expressive, powerful optimizer (e.g., Free-Axis Selection (Frax-is) or Free-Quaternion Selection (FQS)) based on the progress of the cost function, drawing inspiration from early stopping in classical machine learning [65] [67].
Two primary switching criteria have been developed:
- Patience-Based Criterion: The optimizer switches when the cost function fails to improve by a threshold ( \epsilon ) for a consecutive number of iterations (the "patience").
- Moving-Average Criterion: The optimizer switches when the absolute difference between the newest cost value and a moving average of previous values falls below a threshold ( \delta ).
The workflow of this adaptive method is visualized below.
Figure 1: Workflow for early stopping hybrid optimizers that switch between methods based on cost function progress.
These hybrid methods have demonstrated superior performance in terms of convergence speed and final accuracy for various chemical models, including the Heisenberg and Fermi-Hubbard models, compared to static probabilistic or iteration-specific hybrid approaches [65].
Structure Optimization for Quantum Circuits
Beyond parameter optimization, the structure of the PQC itself can be optimized. This involves not just tuning parameters, but also adding or removing gates to find a more expressive and trainable ansatz. A notable protocol involves iteratively growing and compressing the circuit [68]:
- Initialization: Start with a minimal, fixed circuit ansatz.
- Growth Phase: Add a layer of parameterized gates to the circuit.
- Optimization Phase: Optimize the parameters of the expanded circuit using a classical method.
- Compression Phase: Identify and remove gates that contribute minimally to the cost function (e.g., gates whose parameters are near zero), effectively simplifying the circuit.
- Iteration: Repeat steps 2-4 until the performance of the circuit meets the desired threshold.
This approach has been shown to generate shallower circuits that perform significantly better than fixed-structure circuits optimized with parameter updates alone, making it particularly suitable for NISQ devices [68].
Experimental Protocols for Chemistry Applications
Protocol 1: Gibbs Free Energy Profile for Prodrug Activation
The calculation of Gibbs free energy profiles for covalent bond cleavage is a critical task in prodrug design, as it determines whether a reaction proceeds spontaneously under physiological conditions [35].
Table 2: Research Reagent Solutions for Quantum-Enhanced Prodrug Analysis
| Research Reagent / Component | Function in the Protocol |
|---|---|
| Active Space Approximation | Reduces the full molecular system to a manageable few-electron/few-orbital model for simulation on limited-qubit devices [35]. |
| Parity Transformation | Converts the fermionic Hamiltonian of the active space into a qubit Hamiltonian compatible with quantum hardware [35]. |
| Hardware-Efficient ( R_y ) Ansatz | A parameterized quantum circuit with a simple structure (( R_y ) rotations and entangling gates) used in the VQE to prepare the trial molecular wavefunction [35]. |
| Polarizable Continuum Model (PCM) | A solvation model (e.g., ddCOSMO) integrated into the quantum computation to simulate the effect of water in the human body [35]. |
| Readout Error Mitigation | A post-processing technique applied to raw measurement results from the quantum computer to mitigate errors and improve accuracy [35]. |
The detailed workflow is as follows:
- System Preparation: Select key atoms involved in the covalent bond cleavage (e.g., a 2-electron/2-orbital active space for a C–C bond). The remaining electrons are treated with a classical method like Hartree-Fock.
- Hamiltonian Generation: Generate the fermionic Hamiltonian within the active space and the chosen basis set (e.g., 6-311G(d,p)). Transform it into a qubit Hamiltonian using a mapping such as parity transformation.
- VQE Execution: a. Circuit Preparation: Initialize a hardware-efficient PQC. b. Energy Estimation: Measure the expectation value of the qubit Hamiltonian. c. Classical Optimization: Use a classical optimizer (e.g., a hybrid method) to minimize the energy expectation value.
- Solvation Energy Calculation: Integrate a solvation model like PCM into the VQE loop to compute the solvent-influenced single-point energies along the reaction path.
- Thermal Correction: Calculate thermal and zero-point energy corrections at the classical level (e.g., HF) and add them to the VQE-computed electronic energies to obtain the final Gibbs free energy profile.
This protocol has been successfully applied to study the carbon-carbon bond cleavage in the prodrug design for β-lapachone, yielding results consistent with wet lab experiments [35].
Protocol 2: Simulating Drug-Target Covalent Interactions
Quantum computers can enhance Quantum Mechanics/Molecular Mechanics (QM/MM) simulations of drug-target interactions, such as the covalent inhibition of the KRAS G12C protein by Sotorasib [35]. The protocol focuses on computing accurate forces for the QM region.
- System Partitioning: Divide the full drug-protein complex into a QM region (the covalent binding site and the inhibitor's reactive moiety) and an MM region (the rest of the protein and solvent).
- Force Calculation via Hellmann-Feynman Theorem: Use the optimized wavefunction from a VQE simulation of the QM region to compute the forces on the nuclei. This involves evaluating the derivative of the electronic Hamiltonian with respect to nuclear coordinates.
- Molecular Dynamics (MD) Propagation: Use the computed quantum forces to propagate the dynamics of the atoms in the QM region, while the MM region is handled with classical force fields.
- Analysis: Analyze the simulation trajectory to quantify bonding interactions, energy profiles, and binding affinities, providing atomic-level insight into the drug's mechanism of action.
The overall logical relationship and data flow between the classical computer, quantum computer, and the target chemical problem in a VQA is summarized below.
Figure 2: High-level logical dataflow of a Variational Quantum Algorithm (VQA) for chemical problems.
Hardware Considerations and the Path Forward
The performance of all optimization techniques is intrinsically linked to the quality of underlying quantum hardware. Key metrics such as qubit coherence time—the duration for which quantum information remains intact—directly constrain the depth and complexity of executable PQCs. Recent material science breakthroughs, such as the development of 2D transmon qubits using tantalum on silicon substrates, have achieved coherence times exceeding 1 millisecond, a nearly 15-fold improvement over industry standards [69]. Such advancements are crucial for enabling the deeper circuits required for complex chemistry simulations and for implementing efficient quantum error correction, which is necessary for fault-tolerant quantum computation. As hardware continues to mature, optimizing the intricate interplay between algorithm design, parameter optimization strategies, and physical device capabilities will remain a central research focus for achieving quantum utility in combinatorial chemistry.
Managing Noise and Decoherence in NISQ-Era Quantum Simulations
The advent of Noisy Intermediate-Scale Quantum (NISQ) computing has opened new pathways for tackling complex problems in combinatorial chemistry research, particularly through Variational Quantum Algorithms (VQAs) like the Variational Quantum Eigensolver (VQE) [1]. These hybrid quantum-classical algorithms are specifically designed to operate on quantum processors containing up to several hundred qubits that are sensitive to their environment and prone to decoherence [1]. For quantum simulations in chemistry, the central challenge lies in maintaining the integrity of quantum information long enough to perform meaningful computations, despite the persistent presence of noise and decoherence that characterize current-generation hardware [70] [71].
This technical guide examines the core strategies for managing these limitations within the context of quantum simulations for combinatorial chemistry. We explore the fundamental nature of quantum errors, systematic characterization methods, and a layered framework of mitigation techniques that range from circuit-level optimizations to advanced post-processing algorithms. By implementing these protocols, researchers can extract more reliable chemical predictions from today's imperfect quantum hardware, advancing the frontier of quantum-accelerated drug discovery and materials design.
Understanding Noise and Decoherence in Quantum Hardware
In quantum computing, decoherence is the process by which a quantum system loses its quantum behavior and begins to behave classically due to interactions with its environment [71]. This phenomenon directly undermines the superposition and entanglement that power quantum algorithms [70]. The primary sources of error in NISQ devices include:
- Environmental Interactions: Qubits are extremely sensitive to their surroundings. Interactions with external particles—including photons, phonons, or magnetic fields—can disturb the quantum state, effectively "measuring" the system and collapsing the wave function [71].
- Imperfect Control Signals: Noisy control pulses used to manipulate qubits can introduce unwanted transitions and distortions in quantum operations [71].
- Material Defects: Microscopic material imperfections create localized charge or magnetic fluctuations that disturb qubit behavior [71].
- Crosstalk: Operations on one qubit may unintentionally affect neighboring qubits [72].
Operational Impact on Quantum Simulations
The cumulative effect of these error sources manifests in several critical limitations for quantum simulations:
- Limited Circuit Depth: Decoherence introduces noise that collapses quantum states prematurely, restricting the number of sequential operations (circuit depth) that can be performed before calculations become unreliable [71]. This directly limits the complexity of quantum circuits that can be executed successfully.
- Gate Infidelities: Current NISQ devices typically achieve single-qubit gate fidelities around 99-99.5% and two-qubit gate fidelities between 95-99% [1]. While impressive, these error rates accumulate rapidly in circuits with thousands of operations.
- Measurement Errors: The process of reading out qubit states is itself prone to errors, where the true "0" or "1" state may be misreported [73].
Table 1: Characteristic Error Rates and Coherence Times in Modern NISQ Processors
| Hardware Platform | Qubit Count | Typical T₁/T₂ times (μs) | Single-Qubit Gate Fidelity | Two-Qubit Gate Fidelity |
|---|---|---|---|---|
| IBM Heron R3 | 136 | ~200-300 [71] | >99.9% [29] | >99.9% (best links) [29] |
| Quantinuum H-Series | ~10,000 [1] | >99.9% [1] | 99.9% [1] | |
| Typical Superconducting | 50-1000 | ~100 [1] | 99-99.5% [1] | 95-99% [1] |
Characterizing and Quantifying Errors
Effective error management begins with precise characterization of noise profiles and error rates. For chemical simulations using VQEs, understanding how errors affect the measured energy expectation values is crucial for assessing result credibility.
The Qubit Error Probability (QEP) Metric
A recent advancement in error characterization is the Qubit Error Probability (QEP) metric, which estimates the probability that an individual qubit will experience an error during computation [72]. Unlike total error probabilities that treat the entire quantum circuit as a monolithic entity, QEP provides a more granular view by focusing on individual qubits, enabling more targeted and effective error mitigation strategies.
The QEP can be incorporated into existing frameworks like the Tool for Error Description in Quantum Circuits (TED-qc), which calculates error probabilities based on hardware calibration data without requiring circuit execution [72]. This pre-processing approach helps researchers predict circuit reliability before consuming valuable quantum compute resources.
Experimental Protocol for Error Characterization
For researchers implementing VQE for molecular systems, we recommend the following characterization protocol:
Baseline Calibration: For each qubit in your chosen processor, extract the latest relaxation time (T₁), dephasing time (T₂), single-qubit gate error, two-qubit gate error, and readout error from the provider's calibration data.
Circuit-Specific QEP Calculation: Using the TED-qc methodology or similar tools, compute the QEP for each qubit in your intended quantum circuit topology, accounting for the specific gate sequence and duration [72].
Symmetry Verification: For chemistry problems, identify and measure conserved quantities (e.g., particle number, total spin) to detect when errors have driven the state outside the physically meaningful subspace [73].
Error Amplification Tests: Intentionally run circuits with amplified noise (e.g., through pulse stretching or gate repetition) to establish how errors scale with circuit depth and complexity [72].
Error Mitigation Techniques and Experimental Protocols
Error mitigation techniques operate at the software and post-processing level to reduce the impact of noise without requiring the extensive qubit overhead of full quantum error correction [73]. These methods are essential for extracting chemically accurate results from NISQ devices.
Core Error Mitigation Methods
Table 2: Error Mitigation Techniques for Quantum Chemistry Simulations
| Technique | Mechanism | Classical Overhead | Best-Suited Application |
|---|---|---|---|
| Zero-Noise Extrapolation (ZNE) | Artificially amplifies noise and extrapolates back to zero noise [73] | Low to Moderate | Ground state energy calculations [72] |
| Probabilistic Error Cancellation (PEC) | Applies inverse noise operations with sampling [73] | High (exponential in error rate) | Precise expectation value measurement |
| Symmetry Verification | Post-selects results obeying physical symmetries [73] | Moderate (scales with subspace size) | Quantum chemistry simulations [73] |
| Measurement Error Mitigation | Builds confusion matrix to correct readout errors [73] | Low | All circuit measurements |
| Zero Error Probability Extrapolation (ZEPE) | Uses QEP metric for more accurate ZNE [72] | Low to Moderate | Mid-depth circuits [72] |
Detailed Experimental Protocols
Zero-Noise Extrapolation (ZNE) with Qubit Error Probability
The standard ZNE approach can be enhanced by incorporating the QEP metric, creating a more physically accurate extrapolation method called Zero Error Probability Extrapolation (ZEPE) [72]:
Circuit Preparation: Implement your VQE ansatz circuit for the target molecular Hamiltonian.
Noise Scaling: Instead of simply repeating gate sequences, scale noise in a physically informed way using the QEP metric. This can be achieved through:
- Pulse stretching to increase gate durations
- Insertion of identity gates or gate pairs that ideally cancel
- Adjusting parameters based on actual QEP values [72]
Execution: Run the circuit at multiple noise scaling factors (e.g., 1×, 2×, 3× the base error level).
Extrapolation: Fit the measured molecular energy values against the mean QEP (not just an abstract scaling factor) and extrapolate to zero error probability [72].
This QEP-enhanced approach has demonstrated superior performance compared to standard ZNE, particularly for mid-depth circuits where error scaling becomes non-linear [72].
Symmetry Verification for Molecular Simulations
For quantum chemistry problems, symmetry verification leverages conserved physical quantities to detect and correct errors:
Identify Conserved Quantities: Determine the symmetries of your molecular Hamiltonian, such as particle number (for fermionic systems) or total spin symmetry.
Circuit Modification: Add measurements for the symmetry operators without disturbing the primary energy estimation.
Post-Selection: Discard results that violate the known symmetries, or re-weight the results based on symmetry compliance [73].
Subspace Expansion: For advanced implementations, use techniques like Quantum Subspace Expansion (QSE) to project results back into the correct symmetry subspace [73].
This approach has proven particularly effective for VQE simulations of small molecules, enabling results with chemical accuracy (within 1 kcal/mol) even on noisy devices [73].
Advanced Noise-Adaptive Algorithms and Circuit Optimization
Noise-Adaptive Quantum Algorithms (NAQAs)
A paradigm shift in NISQ algorithm design involves treating noise not just as a detriment to be mitigated, but as a feature that can be exploited. Noise-Adaptive Quantum Algorithms (NAQAs) leverage multiple noisy outputs to guide the optimization process [74].
The core NAQA framework operates through an iterative process:
- Sample Generation: Obtain multiple samples from a noisy quantum circuit (e.g., VQE or QAOA).
- Problem Adaptation: Analyze the noisy samples to identify patterns or "attractor states" that guide the optimization.
- Re-optimization: Adjust the problem Hamiltonian or circuit parameters based on the noise-derived insights.
- Iteration: Repeat until solution quality converges [74].
This approach bears conceptual resemblance to classical Cross-Entropy Methods but specifically exploits quantum correlations in noisy outputs. Empirical results suggest NAQAs can outperform standard VQE and QAOA in noisy environments, though with increased computational overhead [74].
Circuit Parallelization and Cutting Techniques
For problems exceeding available qubit counts, circuit cutting and parallelization techniques enable the distribution of large quantum circuits across multiple smaller quantum processors or sequential executions on a single device [6].
The fundamental approach involves:
- Problem Decomposition: Identify naturally separable subproblems within the larger quantum chemistry simulation.
- Slice Implementation: Implement each subproblem as an independent, smaller parameterized quantum circuit (slice).
- Parallel Execution: Execute slices simultaneously on available hardware.
- Result Reconstruction: Combine results through classical post-processing to approximate the solution to the original problem [6].
While this introduces an exponential overhead in the number of measurements required for exact reconstruction, careful problem selection can minimize this penalty. For combinatorial chemistry problems with inherent locality, the approximation can remain highly accurate while dramatically reducing quantum resource requirements [6].
Dynamic Circuits and Active Reset
Advanced control capabilities enable dynamic circuits that incorporate classical processing mid-computation. These techniques can significantly reduce circuit depth and improve accuracy:
- Mid-Circuit Measurement: Measure qubits during circuit execution rather than only at the end.
- Feed-Forward Operations: Use measurement results to conditionally alter subsequent operations.
- Active Qubit Reset: Re-initialize measured qubits to the ground state for reuse in the same circuit [29].
Recent demonstrations on IBM hardware have shown dynamic circuits can provide up to 25% more accurate results with a 58% reduction in two-qubit gates for a 46-site Ising model simulation with 8 Trotter steps [29].
Table 3: Key Experimental Resources for Noise-Resilient Quantum Chemistry Simulations
| Resource Category | Specific Tools/Solutions | Function/Purpose |
|---|---|---|
| Error Mitigation Frameworks | Mitiq, Qiskit Runtime | Software implementations of ZNE, PEC, and measurement mitigation [73] |
| Hardware Access | IBM Quantum Heron R3, Quantinuum H-Series | High-fidelity processors with advanced control capabilities [29] |
| Circuit Optimization Tools | Qiskit Transpiler, TKet | Optimize quantum circuits for specific hardware topology and noise profiles |
| Characterization Tools | TED-qc, QEP Calculator | Pre-execution error prediction and circuit reliability assessment [72] |
| Specialized Functions | Qiskit Functions Catalog | Pre-built, optimized circuits for common quantum chemistry applications [29] |
Workflow and System Architecture
The following diagram illustrates the integrated workflow for managing noise and decoherence in quantum chemistry simulations, incorporating the techniques discussed throughout this guide:
Noise-Adaptive Workflow for Quantum Chemistry
Managing noise and decoherence in NISQ-era quantum simulations requires a comprehensive, layered strategy that spans from fundamental hardware characterization to advanced algorithmic techniques. By implementing the protocols outlined in this guide—including QEP-enhanced error mitigation, symmetry verification, noise-adaptive algorithms, and circuit optimization—researchers can significantly improve the reliability of quantum simulations for combinatorial chemistry applications.
As quantum hardware continues to evolve with processors like IBM's Nighthawk and Quantinuum's H-series pushing the boundaries of fidelity and scale [29], these error management techniques will remain essential for bridging the gap between current capabilities and the future potential of fault-tolerant quantum computation. The iterative co-design of algorithms and hardware, informed by a deep understanding of noise and decoherence, will ultimately enable quantum computers to fulfill their promise in accelerating drug discovery and materials science.
Hamiltonian Scaling and other Techniques for Accelerated Convergence
Variational Quantum Algorithms (VQAs) represent a promising class of hybrid quantum-classical approaches for leveraging contemporary Noisy Intermediate-Scale Quantum (NISQ) devices. Their application to combinatorial chemistry research, particularly in simulating molecular systems and optimizing chemical processes, offers potential pathways to quantum advantage. A critical component governing the performance of these algorithms is the Hamiltonian, which defines the system's energy landscape and physical properties. The efficiency of VQAs is heavily influenced by how the underlying Hamiltonian scales with system complexity and the techniques employed to accelerate algorithmic convergence. Within the broader thesis on applying VQAs to combinatorial chemistry, this guide provides an in-depth technical examination of Hamiltonian scaling properties and advanced methods for achieving faster, more reliable convergence in quantum simulations. The focus on accelerated convergence is paramount for making these algorithms practical for drug development professionals and researchers who require timely and accurate results for molecular design and analysis.
The document is structured as follows. First, it details the scaling challenges associated with electronic structure Hamiltonians in chemical simulations. Subsequently, it explores distributed optimization schemes and performance comparison frameworks designed to mitigate these challenges. Finally, it outlines specific experimental protocols and the essential toolkit required for practical implementation.
Hamiltonian Scaling in Electronic Structure Problems
The electronic structure Hamiltonian is central to quantum chemistry calculations, as it encodes the interactions between electrons and nuclei in a molecule. The dimension of this Hamiltonian scales with the size of the basis set used to discretize the wavefunction, presenting a fundamental challenge for simulation.
When a chemical system with 𝑁 electrons is modeled using a basis set 𝑆={𝜙𝑖} containing 𝐾 basis functions, the wavefunction is represented as a 𝐾×1 column vector. Consequently, the Hamiltonian takes the form of a 𝐾×𝐾 matrix [75]. The primary scalability issue stems from the relationship between the number of basis functions (𝐾) and the number of electrons or atoms. For precise calculations, 𝐾 must increase to capture more complex electron correlations, leading to an exponential growth in the Hamiltonian's dimension. This exponential scaling makes the problem intractable for classical computers as molecule size increases, which is the key motivation for using quantum computers.
Table: Scaling Relationships for Electronic Structure Hamiltonians
| System Parameter | Symbol | Impact on Hamiltonian | Scaling Characteristic |
|---|---|---|---|
| Number of Basis Functions | 𝐾 | Determines matrix dimension | 𝐾×𝐾 matrix |
| Number of Electrons | 𝑁 | Influences required 𝐾 | Exponential in worst case |
| Classical Diagonalization | - | Computational time | 𝑂(𝑁³) to 𝑂(𝑁²‧³⁷) |
For quantum algorithms like the Variational Quantum Eigensolver (VQE), which aim to find the ground state energy of such Hamiltonians, this scaling directly impacts the quantum circuit requirements. The number of qubits must be sufficient to represent the Hamiltonian, and the circuit depth (number of gates) is influenced by the Hamiltonian's complexity, such as the number of non-commuting terms that require separate measurement [76] [77] [78].
Techniques for Accelerated Convergence
Distributed Optimization Schemes
A prominent approach to accelerate VQAs is through distributed optimization. The QUDIO (Quantum Distributed Optimization) scheme addresses the computational overhead of VQAs by partitioning the learning problem across multiple quantum processors [79]. In this framework, a classical central server divides the overall problem into smaller subproblems. Each subproblem is allocated to a local node, which consists of a quantum processor and a classical optimizer. These local nodes then perform parallel optimization on their respective subproblems. A key feature of QUDIO is the timely synchronization of optimization information (e.g., parameter updates) across all local nodes by the central server. This synchronous exchange ensures that the distributed optimization remains coherent and converges towards a global solution. The authors prove a sublinear convergence rate for QUDIO under ideal conditions and report numerical results demonstrating a superlinear runtime speedup with respect to the number of local nodes [79].
Performance Comparison and Toolchain Portability
Another technique for enhancing performance involves systematic comparison and toolchain portability across different high-performance computing (HPC) systems. A generic problem description format, encompassing the Hamiltonian and the variational ansatz, allows for consistent porting of a problem definition among different quantum simulators [76]. This approach enables researchers to study the dependence of performance on the runtime environment, the scalability of various simulation codes, and the mutual agreement of physical results. The use of a unified toolchain helps identify the most efficient simulator and configuration for a specific problem, indirectly accelerating convergence by selecting the optimal computational environment. Furthermore, techniques like job arrays can be employed to mitigate the limited parallelism inherent in variational algorithms, which are often constrained by long runtimes relative to their memory footprint [76].
Algorithmic and Optimizer Enhancements
Convergence can also be accelerated through algorithmic improvements and the choice of classical optimizers. Comparative studies have shown that variational methods, such as VQE, are significantly less demanding in terms of quantum circuit depth and the number of two-qubit gates compared to adiabatic approaches [77]. This resource efficiency reduces noise susceptibility and facilitates faster iteration. To address the potential for slow classical optimization, strategies for initializing the parameters of the variational circuit have been developed. Providing a good initial guess, potentially derived from approximate classical methods or problem structure, can place the optimizer closer to the optimum, thereby significantly speeding up the convergence process [77]. Additionally, the choice between gradient-based and gradient-free optimizers can impact convergence speed, especially in the presence of experimental noise [80].
Table: Acceleration Techniques for Variational Quantum Algorithms
| Technique | Mechanism | Key Advantage | Considerations |
|---|---|---|---|
| QUDIO [79] | Distributed parallel optimization across quantum processors | Superlinear runtime speedup | Requires multiple quantum processors; synchronization overhead |
| Unified Toolchain [76] | Consistent problem porting across HPC simulators | Identifies optimal runtime environment | Limited by intrinsic VQA parallelism |
| Advanced Optimizers [77] [80] | Improved initial parameter guess; gradient rules | Reduces number of optimization cycles | Performance is problem and noise-dependent |
Experimental Protocols for Validation
Protocol for Distributed Optimization (QUDIO)
- Problem Partitioning: A classical central server divides the full VQA optimization problem into 𝑁 distinct subproblems.
- Node Allocation: Each subproblem is assigned to a local node 𝑖, which comprises a quantum processor and a classical optimizer (e.g., ADAM, SPSA).
- Parallel Optimization Loop: For a predefined number of global epochs: a. Local Computation: Each node 𝑖 runs its quantum circuit and classical optimizer on its local subproblem for a fixed number of steps. b. Synchronization: The central server collects the parameter vectors (e.g., 𝜃₁, 𝜃₂, ..., 𝜃N) from all local nodes. c. Parameter Averaging: The server computes the average parameter vector, 𝜃̄ = (1/𝑁) ∑ 𝜃𝑖. d. Broadcast: The server broadcasts the averaged parameter vector 𝜃̄ back to all local nodes. e. Parameter Update: Each local node 𝑖 sets its local parameters to the received averaged value, 𝜃_𝑖 = 𝜃̄, ensuring synchronization.
- Validation: The final synchronized parameters are used to evaluate the global cost function on the full problem to assess convergence and solution quality [79].
Protocol for Performance Benchmarking on HPC
- Problem Definition: Formulate the problem (e.g., H₂ ground state, MaxCut) using a generic, simulator-agnostic description of the Hamiltonian and ansatz.
- Toolchain Translation: Employ a parser tool to automatically translate the problem definition into the native input format for a set of target quantum simulators (e.g., Qiskit, Cirq, PennyLane).
- HPC Execution: Run the translated problems on a selection of HPC systems, ensuring consistent resource allocation (e.g., node count, memory) where possible.
- Data Collection: For each run, record key performance metrics: total wall-clock time, time-to-solution, memory footprint, and the final physical result (e.g., ground state energy).
- Comparative Analysis: Analyze the collected data to rank simulator performance, assess scalability with problem size, and verify that all simulators produce mutually agreeable physical results within a defined tolerance [76].
Protocol for Metrology Optimization
- Setup: Prepare a displaced squeezed state as a probe, with fixed squeezing (𝑟) and displacement (∣𝛼∣) but a tunable displacement phase angle (𝜙_𝛼).
- Parameter Encoding: Apply a small, fixed phase shift (the parameter to be sensed) to the probe state.
- Measurement: Perform homodyne detection with a tunable detection angle (𝜙_𝐻𝐷).
- Cost Calculation: Estimate the classical Fisher Information ℱ(𝜙𝛼, 𝜙𝐻𝐷) from measurement statistics. The cost function is its inverse: 𝐶(𝜙𝛼, 𝜙𝐻𝐷)=1/ℱ.
- Optimization: a. Gradient-Based: Use Gaussian parameter-shift rules to estimate the gradient of 𝐶 directly from measurements. Update parameters via gradient descent. b. Gradient-Free: Feed the cost function 𝐶 directly to a Bayesian optimizer for fine-tuning.
- Iteration: Repeat steps 1-5, updating (𝜙𝛼, 𝜙𝐻𝐷) based on the optimizer's output until convergence is achieved [80].
The Scientist's Toolkit: Research Reagent Solutions
The experimental implementation of accelerated VQAs, particularly in quantum sensing and metrology, relies on a set of critical physical components and classical algorithms.
Table: Essential Research Reagents for Variational Metrology Experiments
| Reagent / Tool | Type | Function in Experiment |
|---|---|---|
| Optical Parametric Oscillator (OPO) | Physical Hardware | Generates squeezed vacuum states, a non-classical resource for enhancing measurement precision [80]. |
| Homodyne Detector | Physical Hardware | Measures the quadratures of the quantum optical field, providing the data for Fisher information calculation [80]. |
| Gaussian Parameter-Shift Rules | Algorithmic Tool | Enables the calculation of gradients of the cost function directly from quantum measurements, facilitating gradient-based optimization [80]. |
| Bayesian Optimizer | Classical Software | A gradient-free optimization algorithm used to fine-tune parameters by modeling the cost function, effective in noisy environments [80]. |
| Classical Fisher Information | Analytical Metric | Quantifies the precision of a measurement protocol; its inverse is used as the cost function to be minimized [80]. |
Benchmarking VQA Performance: Validation and Comparison with Classical Methods
Variational Quantum Algorithms (VQAs) have emerged as a promising framework for leveraging current noisy intermediate-scale quantum (NISQ) processors to tackle problems in quantum chemistry and combinatorial optimization [6]. These hybrid quantum-classical algorithms use a parameterized quantum circuit as a black-box function optimized by a classical computer, aiming to find ground states of molecular systems or solutions to optimization problems [6]. As VQAs develop, establishing rigorous benchmarks against established classical computational chemistry methods becomes essential for assessing their current utility and guiding future development.
This technical guide provides a structured approach for comparing VQA performance against three cornerstone classical methods: Hartree-Fock (HF), Density Functional Theory (DFT), and Complete Active Space Configuration Interaction (CASCI). We present quantitative benchmarking data, detailed experimental protocols, and standardized workflows to enable researchers to conduct systematic evaluations of algorithmic performance across chemical systems of increasing complexity.
Classical Methods Hierarchy
Classical computational chemistry methods form a multi-tiered hierarchy of increasing accuracy and computational cost. The Hartree-Fock (HF) method provides a foundational wavefunction-based approach that approximates the many-electron wavefunction as a single Slater determinant [81]. HF employs the self-consistent field (SCF) procedure to optimize orbitals but neglects electron correlation except for the exchange interaction accounted for via the antisymmetry principle [81].
Density Functional Theory (DFT) bypasses the wavefunction entirely, using electron density as the fundamental variable [82]. According to the Hohenberg-Kohn theorems, all ground-state properties are uniquely determined by the electron density [82]. Modern DFT employs the Kohn-Sham scheme to map the interacting system onto a fictitious non-interacting system with the same density, with accuracy critically dependent on the approximate exchange-correlation functional used [82] [83].
Complete Active Space (CAS) methods, including CASSCF and CASCI, explicitly handle multireference character and static correlation by performing a full configuration interaction (CI) calculation within a carefully selected active space of electrons and orbitals [84]. CASSCF variationally optimizes both CI coefficients and molecular orbitals, while CASCI uses a fixed orbital basis [84]. These methods are particularly crucial for describing bond breaking, excited states, and degenerate systems where single-reference methods like HF and DFT fail.
Variational Quantum Algorithms
VQAs encompass algorithms like the Variational Quantum Eigensolver (VQE) and Quantum Approximate Optimization Algorithm (QAOA) [6]. VQE employs a parameterized quantum circuit to prepare trial wavefunctions, whose energy expectation value is measured and minimized by a classical optimizer [6]. The algorithm's success depends on ansatz design, optimization landscapes, and resilience to hardware noise. Recent developments include quantum-selected configuration interaction (QSCI), which uses quantum processors to sample important configurations for subsequent classical diagonalization [85].
Table 1: Core Methodological Characteristics
| Method | Theoretical Foundation | Electron Correlation Treatment | Computational Scaling |
|---|---|---|---|
| Hartree-Fock (HF) | Wavefunction theory (Single Slater determinant) | Neglects electron correlation except Fermi (exchange) | Formal: O(N⁴), Practical: O(N³) |
| Density Functional Theory (DFT) | Electron density via Kohn-Sham equations | Approximate via exchange-correlation functionals | Formal: O(N³), Practical: O(N²-N³) |
| CASCI/CASSCF | Multiconfigurational wavefunction in active space | Exact static correlation within active space | Exponential with active space size |
| VQAs (VQE) | Parameterized quantum circuit (variational principle) | Depends on ansatz expressibility | Quantum circuit: depends on ansatz; Classical: parameter optimization |
Benchmarking Metrics and Performance Data
Quantitative Accuracy Assessment
Recent comprehensive benchmarking provides critical insights into the relative performance of computational methods. The Gold-Standard Chemical Database 138 (GSCDB138), a rigorously curated benchmark library, offers high-accuracy reference data across diverse chemical systems [83]. This database includes 138 datasets (8,383 entries) covering main-group and transition-metal reaction energies, barrier heights, non-covalent interactions, and molecular properties, with reference values primarily from coupled cluster theory [83].
DFT performance varies significantly with functional choice. Meta-GGA functionals like r²SCAN-D4 rival hybrid functionals for vibrational frequencies, while B97M-V and ωB97X-V emerge as the most balanced hybrid meta-GGA and hybrid GGA functionals, respectively [83]. Double hybrid functionals lower mean errors by approximately 25% compared to the best hybrids but demand careful treatment of frozen-core approximations, basis sets, and multireference character [83].
For VQAs applied to combinatorial optimization, studies on problems like the Multiple Knapsack Problem show that variational quantum imaginary time evolution (VarQITE) can achieve significantly lower mean optimality gaps compared to QAOA and other conventional methods [5]. Furthermore, scaling Hamiltonian coefficients can reduce optimization costs and accelerate convergence [5].
Table 2: Representative Performance Benchmarks Across Chemical Properties (Mean Absolute Errors)
| Property Class | Representative System | HF | B3LYP (DFT) | ωB97X-V (DFT) | CASSCF | VQE/VQA |
|---|---|---|---|---|---|---|
| Barrier Heights (kcal/mol) | Hydrogen transfer reactions | 15.2 | 4.3 | 2.1 | 6.8 | Varies significantly with ansatz |
| Reaction Energies (kcal/mol) | Main-group thermochemistry | 20.5 | 3.8 | 1.9 | 12.4 | Limited data |
| Non-covalent Interactions (kcal/mol) | S22 dataset | 4.2 | 1.3 | 0.7 | 2.5 | Highly system-dependent |
| Dipole Moments (D) | Small polar molecules | 0.35 | 0.12 | 0.08 | 0.28 | Circuit depth limited |
| Vibrational Frequencies (cm⁻¹) | Small molecular dimers | 150 | 40 | 25 | 90 | Emerging capability |
Resource Requirements and Scalability
Computational resource requirements differ substantially across methods. HF and DFT demonstrate polynomial scaling with system size (typically O(N³) to O(N⁴)), making them applicable to medium and large systems [81] [82]. CASSCF faces exponential scaling with active space size, currently limited to approximately 18 electrons in 18 orbitals in conventional implementations [84].
VQAs face distinct resource constraints: qubit count, circuit depth, and measurement statistics. Current hardware limitations restrict VQA implementations to small toy instances of industrially relevant problems [6]. For example, parallelization frameworks that split quantum circuits allow problems larger than available qubits to be addressed, but with potential information loss [6]. Compact wavefunction representations, such as those achieved through quantum-selected configuration interaction, can reduce configuration space by over 200 times compared to conventional SCI selection for strongly correlated systems [85].
Experimental Protocols for Method Comparison
Standardized Benchmarking Workflow
Method-Specific Configuration Protocols
Hartree-Fock Protocol
HF calculations begin with defining the molecular geometry and basis set. The self-consistent field procedure iteratively solves the Roothaan-Hall equations until convergence criteria are met (typically energy change < 10⁻⁶ Hartree and density change < 10⁻⁵) [81]. For closed-shell systems, use restricted HF (RHF); for open-shell, unrestricted HF (UHF) or restricted open-shell HF (ROHF). The HF method provides a reference wavefunction for post-HF methods but exhibits systematic errors due to neglected electron correlation, particularly severe for reaction energies, barrier heights, and dispersion interactions [81].
DFT Implementation Protocol
Select an exchange-correlation functional appropriate for the target properties. For general-purpose applications, hybrid functionals like ωB97X-V or meta-GGAs like B97M-V offer balanced performance [83]. Employ a triple-zeta quality basis set with polarization functions (e.g., def2-TZVP). For periodic systems, use plane-wave basis sets with appropriate pseudopotentials. Perform numerical integration with fine grids (e.g., 75–100 radial points, 302 angular points). Conduct structural optimization until forces are below 0.001 Hartree/Bohr. DFT excels for ground-state properties of medium to large systems but struggles with charge transfer, dispersion, and strongly correlated systems [82] [83].
CASCI/CASSCF Implementation Protocol
The critical step is active space selection. Identify the orbitals and electrons directly involved in the chemical process of interest. For organic diradicals, this typically includes the frontier orbitals; for transition metal complexes, include metal d-orbitals and ligand donor orbitals [84]. Perform initial HF calculation to generate starting orbitals, then transform to appropriate orbitals (natural, localized, or pseudo-canonical). For state-averaged calculations, assign equal weights to states of interest. CASSCF optimizes both orbitals and CI coefficients; CASCI uses fixed orbitals. These methods provide qualitative correct description of multireference systems but miss dynamic correlation [84].
VQA Experimental Protocol
For VQE, map the electronic structure problem to a qubit Hamiltonian using Jordan-Wigner or Bravyi-Kitaev transformation. Prepare a parameterized ansatz—either problem-inspired (UCCSD) or hardware-efficient. Measure the energy expectation value through repeated circuit execution. Employ classical optimizers (BFGS, SPSA, or gradient-free methods) to minimize energy [6]. For combinatorial problems using QAOA, design phase and mixer operators reflecting problem constraints [5]. Mitigate hardware errors using readout error mitigation, zero-noise extrapolation, and dynamical decoupling. VQA performance heavily depends on ansatz expressibility, optimizer choice, and error mitigation strategies [6].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools and Resources
| Tool Category | Specific Tools/Resources | Primary Function | Application Context |
|---|---|---|---|
| Reference Databases | GSCDB138 [83], GMTKN55 [83] | Provide gold-standard reference data | Method validation and functional development |
| Quantum Chemistry Packages | PySCF, Q-Chem, Gaussian, ORCA | Implement HF, DFT, CASSCF calculations | Classical method reference calculations |
| Quantum Development Frameworks | Qiskit, Cirq, PennyLane | VQA implementation and simulation | Quantum algorithm development |
| Basis Sets | cc-pVXZ, def2-XZVP, 6-31G* [84] | Define atomic orbital basis functions | All electronic structure methods |
| Active Space Selection Tools | AVAS, AOIM, automatic active space selection | Identify important orbitals for CAS | Multireference calculations |
| Error Mitigation Techniques | Zero-noise extrapolation, readout mitigation | Reduce hardware errors in VQAs | NISQ-era quantum computations |
Comparative Analysis and Interpretation Guidelines
Method Selection Framework
Interpretation and Error Analysis
When analyzing benchmarking results, distinguish between different error types: methodological approximations, basis set incompleteness, active space selection, and quantum hardware noise. For VQAs, particular attention should be paid to the optimization landscape, as VQAs are hindered by issues such as barren plateaus and suboptimal local minima [5].
Systematic error patterns emerge across methods. HF consistently overestimates reaction barriers and binding energies due to neglect of electron correlation [81]. DFT errors vary with functional: GGA functionals underestimate barrier heights, while hybrids provide better performance [83]. CASSCF captures static correlation but misses dynamic correlation, leading to underestimated binding energies [84]. VQA errors are more variable, depending on ansatz expressibility, optimizer performance, and hardware noise [6].
For transition metal systems and non-covalent interactions, recommend using double-hybrid DFT or CASSCF with post-dynamical correlation treatment (e.g., CASPT2 or MRCI) for highest accuracy [83]. For large systems where high-level methods are prohibitive, well-validated DFT functionals like ωB97X-V or B97M-V provide the best compromise [83].
Benchmarking VQAs against established classical computational chemistry methods reveals a complex landscape where no single approach dominates across all chemical systems and properties. HF provides a qualitative foundation but lacks quantitative accuracy. DFT offers the best balance of efficiency and accuracy for single-reference systems. CAS methods excel for multireference problems but face exponential scaling. VQAs show promise for specific applications like combinatorial optimization and strong correlation but remain limited by hardware constraints and optimization challenges.
As quantum hardware advances, the most productive path forward likely involves hybrid quantum-classical approaches, such as using quantum processors for state sampling in QSCI [85] or combining efficient classical geometry optimization with targeted quantum energy evaluation [86]. The benchmarking frameworks and protocols established here provide researchers with the tools to rigorously assess progress toward practical quantum advantage in computational chemistry.
Variational Quantum Algorithms (VQAs) represent a promising class of hybrid quantum-classical algorithms designed to leverage contemporary Noisy Intermediate-Scale Quantum (NISQ) devices for solving classically intractable problems in fields such as combinatorial chemistry and optimization [2]. These algorithms, including the Variational Quantum Eigensolver (VQE) and the Quantum Approximate Optimization Algorithm (QAOA), operate by optimizing parameterized quantum circuits through a classical feedback loop [2]. The performance of VQAs is fundamentally governed by three interconnected metrics: accuracy, which reflects the solution quality relative to the true ground state energy; convergence, which characterizes the algorithmic trajectory toward a final solution; and time-to-solution, which encompasses the total computational resources required [87] [2]. For researchers in drug development and combinatorial chemistry, where identifying molecular ground states is crucial, a deep understanding of these metrics is essential for evaluating the potential quantum advantage offered by VQAs. This guide provides a technical examination of these performance metrics, grounded in recent benchmarking studies and experimental results.
Core Performance Metrics in VQAs
The evaluation of Variational Quantum Algorithms hinges on quantifiable metrics that determine their practical utility and efficiency. The table below summarizes these core performance metrics and the factors that influence them.
Table 1: Core Performance Metrics for Variational Quantum Algorithms
| Metric | Description | Key Influencing Factors |
|---|---|---|
| Accuracy [87] | Measures solution quality, often via the optimality gap between found and true ground state energy. | Ansatz expressibility, presence of noise, Hamiltonian formulation, and optimization effectiveness. |
| Convergence [64] [2] | The rate and stability with which an algorithm approaches its final solution over iterations. | Optimization landscape (e.g., barren plateaus), classical optimizer choice, and parameter initialization strategies. |
| Time-to-Solution [88] [2] | Total computational time (quantum + classical) required to find a solution within a target accuracy. | Quantum circuit depth, number of measurements (shots), and classical optimization overhead. |
The interplay of these metrics dictates overall algorithmic performance. For instance, a highly accurate ansatz may require a deep circuit, leading to longer execution times on noisy hardware and potentially hindering convergence due to decoherence and gate errors [2]. Furthermore, the phenomenon of barren plateaus—where the gradients of the cost function vanish exponentially with system size—poses a significant challenge to convergence, making it difficult for the optimizer to find a direction for improvement [2]. The choice of the classical optimizer (e.g., gradient-based vs. gradient-free) also critically impacts both convergence behavior and the total time-to-solution, as each evaluation of the cost function requires numerous quantum measurements [2].
Quantitative Benchmarking Data
Recent empirical studies provide critical insights into the practical performance of different VQAs when applied to realistic problems. The following table synthesizes key findings from contemporary research, comparing various algorithms across problem domains and performance metrics.
Table 2: Comparative Performance of Quantum Optimization Algorithms on Benchmark Problems
| Algorithm | Problem Type | Reported Performance | Key Findings |
|---|---|---|---|
| VarQITE [64] | Multiple Knapsack Problem (MKP) | "Significantly lower mean optimality gaps" compared to QAOA and conventional methods. | Hamiltonian coefficient scaling reduces optimization cost and accelerates convergence. |
| VQE/CVaR-VQE [87] | Multi-Dimensional Knapsack (MDKP), Maximum Independent Set (MIS) | Evaluated for "feasibility, optimality gaps, and scalability" in a structured benchmarking framework. | Qubit compression techniques (e.g., PCE, QRAO) help address resource constraints. |
| QAOA [88] | Max-Cut | Identifies a "minimum problem size" needed to consistently outperform classical sampling. | For fixed resources, performance is characterized against greedy algorithms and random sampling. |
| QD-CMA [89] | NP-hard Combinatorial Problems | Shows "superior circuit optimization w.r.t. speed and solution score" versus benchmark algorithms. | Quality Diversity search optimizes circuit architecture, improving efficiency and performance. |
Benchmarking results consistently highlight the trade-offs inherent in VQA performance. For example, while QAOA and its variants are widely applied to combinatorial problems like Max-Cut, their performance for a given resource budget may only surpass simple classical heuristics beyond a certain problem size [88]. In constrained optimization, such as the Multiple Knapsack Problem, algorithms like VarQITE demonstrate a notable advantage in achieving higher accuracy (lower optimality gap) [64]. Furthermore, advanced techniques like qubit compression (e.g., Pauli Correlation Encoding) are being employed to enhance scalability and reduce time-to-solution, which is crucial for applying VQAs to larger, more chemically relevant systems [87].
Experimental Protocols and Methodologies
A rigorous and reproducible experimental protocol is fundamental for a meaningful comparison of VQA performance. This section details a standard workflow for benchmarking VQAs, drawing from established methodologies in recent literature [64] [87].
Diagram 1: VQA Experimental Workflow
Problem Definition and Hamiltonian Formulation
The first step involves mapping the target problem—such as finding the ground state energy of a molecule or solving a combinatorial problem—to a quantum-mechanical Hamiltonian [2]. In quantum chemistry, this is typically the electronic structure Hamiltonian, expressed as a sum of Pauli operators after a fermion-to-qubit mapping (e.g., Jordan-Wigner or Bravyi-Kitaev) [2]. For combinatorial problems, the objective function is encoded into a problem Hamiltonian, often via a Quadratic Unconstrained Binary Optimization (QUBO) formulation. For constrained problems, innovative approaches like the unbalanced QUBO can be used to avoid introducing extra qubits via slack variables, thereby conserving resources [64].
Ansatz Selection and Circuit Construction
The ansatz, or parameterized quantum circuit, defines the search space for the variational algorithm [2]. Common choices include:
- Physically-Inspired Ansätze: Such as the Unitary Coupled Cluster (UCCSD), which is chemically motivated but can lead to deep circuits [2].
- Hardware-Efficient Ansatz (HEA): Uses native gate sets to minimize circuit depth, improving feasibility on NISQ devices but sometimes struggling with trainability [2].
- Quantum Alternating Operator Ansatz (QAOA+): Applies alternating problem and mixer Hamiltonians, and is generalized for constrained problems [64].
- QITE-inspired Ansätze: Such as the imaginary Hamiltonian Variational Ansatz (iHVA), which is tailored for specific problem symmetries [64].
Parameter Initialization and Optimization Loop
Parameters of the ansatz are typically initialized using heuristic strategies to avoid poor local minima [64] [2]. The core of the experiment is the hybrid loop:
- Quantum Execution: The parameterized circuit is executed on a quantum processor (or simulator). The output state is measured repeatedly to estimate the expectation value of the problem Hamiltonian, ⟨H⟩, which serves as the cost function [2].
- Classical Optimization: A classical optimizer uses the measured cost to compute a parameter update. Optimizers range from gradient-based methods (using the parameter-shift rule [2]) to gradient-free algorithms (e.g., COBYLA, SPSA). This loop repeats until a convergence criterion is met, such as a minimal change in the cost function or a maximum number of iterations.
Case Study: VarQITE Protocol for MKP
A specific protocol for applying Variational Quantum Imaginary Time Evolution (VarQITE) to the Multiple Knapsack Problem (MKP) is detailed below [64].
Diagram 2: VarQITE Protocol for MKP
- Problem Encoding: The MKP instance is converted to a QUBO problem. The unbalanced penalty approach is employed to integrate constraints without adding slack variables, thus avoiding an increase in qubit count [64].
- Hamiltonian Conversion: The resulting QUBO formulation is then converted into a Hamiltonian compatible with a Max-Cut problem structure [64].
- Ansatz and Scaling: The imaginary Hamiltonian Variational Ansatz (iHVA), which is specifically designed with bit-flip symmetry for Max-Cut, is constructed. A critical step identified in recent work is the scaling of Hamiltonian coefficients, which has been shown to reduce optimization cost and accelerate convergence [64].
- VarQITE Execution: Instead of a standard classical optimizer, the McLachlan variational principle is used to update the parameters of the iHVA circuit, simulating an evolution in imaginary time. The system is evolved from an initial state over small imaginary time steps Δτ until the energy expectation value converges to a minimum [64].
The Scientist's Toolkit: Key Research Reagents and Materials
The experimental implementation of VQAs relies on a suite of algorithmic "reagents" and computational tools. The following table catalogues essential components for researchers designing VQA experiments in combinatorial chemistry.
Table 3: Essential Research Reagents and Tools for VQA Experimentation
| Tool / Component | Type | Function in the Experiment |
|---|---|---|
| Problem Hamiltonian [2] | Mathematical Formulation | Encodes the target problem (e.g., molecular energy, optimization objective) into a quantum-mechanical operator. |
| Ansatz Circuit [64] [2] | Parameterized Quantum Circuit | Defines the subspace of quantum states explored during optimization (e.g., UCCSD, HEA, QAOA+, iHVA). |
| Classical Optimizer [2] | Algorithm | Adjusts ansatz parameters to minimize the cost function. Choices include gradient-based (e.g., ADAM) and gradient-free (e.g., SPSA) methods. |
| Parameter-Shift Rule [2] | Algorithmic Rule | Enables exact gradient calculation on quantum hardware for specific parameterized gates, crucial for gradient-based optimization. |
| Conditional Value-at-Risk (CVaR) [90] | Statistical Aggregator | Enhances VQE/QAOA performance by focusing the cost function on the best-measured outcomes, improving solution quality. |
| Quantum Random Access Optimization (QRAO) [90] [87] | Qubit Compression Technique | Encodes more classical variables into fewer qubits, improving scalability at the cost of more complex measurements. |
This toolkit enables researchers to construct and deconstruct VQA performance. For example, replacing a standard VQE cost function with a CVaR-based aggregation directly influences the accuracy metric by prioritizing high-quality samples from the measurement statistics [90]. Similarly, employing a qubit-efficient encoding like QRAO directly addresses the time-to-solution metric by reducing the quantum resource requirements for a given problem size, albeit with potential trade-offs in other metrics [87].
Validation through Real-World Case Studies and Experimental Data
Variational Quantum Algorithms (VQAs) represent a class of hybrid quantum-classical algorithms designed to leverage the capabilities of current Noisy Intermediate-Scale Quantum (NISQ) devices. Unlike fully quantum algorithms that require fault-tolerant systems, VQAs use a quantum processor to prepare and measure parameterized quantum states, while a classical optimizer tunes these parameters to minimize a cost function, often the energy of a molecular system [27]. This hybrid approach makes VQAs particularly suitable for the complex computational challenges inherent to combinatorial chemistry research, where the goal is to explore vast chemical spaces to discover new reactions, catalysts, and materials [91].
The core of many chemistry-focused VQAs is the Variational Quantum Eigensolver (VQE), specifically designed to find the ground-state energy of molecular Hamiltonians—a fundamental task in quantum chemistry [27]. The applicability of VQAs extends to optimization problems relevant to chemical synthesis and drug discovery pathways through algorithms like the Quantum Approximate Optimization Algorithm (QAOA) [23] [92]. For researchers in drug development, these algorithms offer a promising path to simulating molecular structures and chemical reactions with an accuracy that is computationally intractable for classical computers alone, potentially shortening development cycles and reducing costs [27].
Core Experimental Protocols for VQAs
The validation of VQAs relies on a consistent and reproducible experimental methodology. The general workflow involves several key building blocks, and its consistency across different software and hardware platforms is critical for obtaining reliable results [92].
General VQA Workflow
The following diagram illustrates the hybrid quantum-classical feedback loop that is fundamental to VQAs like VQE and QAOA.
Detailed Methodological Components
Problem Formulation (Hamiltonian Definition): The chemical system must be translated into a quantum-mechanical description. For molecular simulations, this begins with the electronic Hamiltonian within the Born-Oppenheimer approximation and a selected basis set (e.g., STO-3G). The Hamiltonian is then transformed into a qubit-representable form using techniques like the Jordan-Wigner transformation, resulting in a linear combination of Pauli operators (e.g.,
ZIXI,YYZZ) [92].Ansatz Selection (State Preparation): The parameterized quantum circuit (ansatz) defines the subspace of quantum states explored during the optimization. For quantum chemistry problems, the Unitary Coupled-Cluster Singles and Doubles (UCCSD) ansatz is a popular choice, which is applied to an initial reference state, such as the Hartree-Fock state [92]. For combinatorial problems, QAOA uses an ansatz built by alternating applications of cost and mixer unitaries [23].
Classical Optimization: A classical optimizer is employed to minimize the cost function. A common choice is the gradient-based Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm [92]. The optimizer iteratively suggests new parameters for the quantum circuit, aiming to find the minimum of the cost landscape.
Case Studies and Experimental Data
This section presents empirical data from benchmark studies that validate the performance of VQAs on problems relevant to combinatorial chemistry.
Case Study 1: H2 Molecule Ground State Simulation
The calculation of the ground state energy of a hydrogen molecule is a foundational benchmark for quantum chemistry algorithms [92].
- Algorithm: Variational Quantum Eigensolver (VQE)
- Ansatz: UCCSD
- Qubits: 4 (after Jordan-Wigner transformation)
- Objective: Approximate the exact dissociation curve.
Table 1: Experimental Setup and Results for H2 Simulation
| Metric | Description / Value |
|---|---|
| Molecular System | H2 molecule |
| Basis Set | STO-3G |
| Hamiltonian Transform | Jordan-Wigner |
| Target Observable | Ground State Energy |
| Key Experimental Finding | VQE with UCCSD ansatz successfully reproduced the theoretical dissociation curve for the H2 molecule, validating the algorithm's accuracy for this foundational quantum chemistry problem [92]. |
Case Study 2: MaxCut Problem
The MaxCut problem, a combinatorial optimization challenge, serves as a proxy for complex resource allocation and segmentation problems that can appear in chemical logistics and analysis pipelines [23] [92].
- Algorithm: Quantum Approximate Optimization Algorithm (QAOA)
- Objective: Find a partition of a graph's vertices into two sets that maximizes the number of edges between the sets.
Table 2: Experimental Data for MaxCut using QAOA
| Metric | Description / Value |
|---|---|
| Problem Type | MaxCut on graphs |
| Graph Size | 10 and 20 nodes [23] |
| Circuit Depth | Varied depths (p-layers) tested [23] |
| Key Experimental Finding | Implementation of QAOA demonstrated the algorithm's potential on the MaxCut problem, while also highlighting the challenges associated with parameter optimization and circuit depth as problem scales [23]. The study emphasized QAOA's relevance for near-term quantum devices [92]. |
Performance Comparison on HPC Systems
A 2025 study by De Pascale et al. benchmarked the performance of VQA simulations across different High-Performance Computing (HPC) environments, providing critical data on scalability and computational efficiency [92].
Table 3: Performance Metrics for VQA Simulations on HPC Systems
| Use Case | Key Performance Insight | Scalability Limitation |
|---|---|---|
| H2 Molecule (VQE) | The toolchain successfully translated problem definitions (Hamiltonian & ansatz) consistently across different software simulators, ensuring result agreement [92]. | VQAs are generally limited by long runtimes relative to their memory footprint, exposing limited parallelism to computation [92]. |
| MaxCut (QAOA) | The parser tool enabled consistent performance comparisons and benchmarking across different HPC systems and simulators [92]. | Techniques like job arrays were found to partially mitigate scalability shortcomings by managing multiple parameter evaluations concurrently [92]. |
| Traveling Salesperson Problem (QAOA) | The study confirmed the utility of the developed tools for exploring HPC performance for optimization problems relevant to chemical logistics [92]. | The classical optimization loop remains a computational bottleneck, especially for larger, more complex problem instances [92]. |
The Scientist's Toolkit: Essential Research Reagents
In the context of computational and quantum chemistry, "research reagents" refer to the core algorithmic components and software tools required to conduct experiments with VQAs.
Table 4: Key Research Reagent Solutions for VQA Experiments
| Reagent / Component | Function in the Experimental Workflow | |
|---|---|---|
| Molecular Hamiltonian | The fundamental description of the chemical system's energy, derived in a second quantization formulation and transformed into a qubit-representable form via (e.g., Jordan-Wigner) [92]. | |
| Parameterized Quantum Circuit (Ansatz) | A tunable quantum circuit (e.g., UCCSD for chemistry, QAOA alternating operators for optimization) that prepares the trial quantum state | Ψ(θ)⟩ [92]. |
| Classical Optimizer | An algorithm (e.g., BFGS) that adjusts the parameters (θ) of the quantum circuit to minimize the value of the cost function, driving the system toward the ground state or optimal solution [92]. | |
| High-Performance Computing (HPC) Simulator | Classical software that emulates the execution of quantum circuits on powerful computer clusters, essential for algorithm development and validation before running on physical quantum hardware [91] [92]. | |
| Hamiltonian & Ansatz Parser | A software tool that ensures a consistent problem definition can be ported across different quantum simulators, which is crucial for reproducible and comparable research results [92]. |
Integrated Workflow for Chemical Discovery
The combination of protocols, tools, and algorithms forms a cohesive pipeline for tackling problems in combinatorial chemistry. The workflow for a molecular simulation project, from problem definition to result validation, integrates several of the components previously described.
This end-to-end process enables researchers to take a target molecule from initial definition to the extraction of chemically relevant properties, such as reaction barrier heights or binding affinities, which can inform the design of new drugs and materials [27]. The final validation step against established classical methods is crucial for building confidence in the quantum computational results [92].
Prospects for Quantum Advantage in Combinatorial Chemistry
Combinatorial chemistry, which involves the systematic synthesis and analysis of vast libraries of chemical compounds, faces fundamental computational challenges. Accurately predicting molecular properties, reaction pathways, and catalytic behaviors requires simulating quantum mechanical systems, a task that is intractable for classical computers for all but the smallest molecules. Variational Quantum Algorithms (VQAs) have emerged as a leading near-term approach to address these challenges by leveraging the synergistic capabilities of quantum and classical computers [12].
These hybrid algorithms are particularly suited for today's Noisy Intermediate-Scale Quantum (NISQ) hardware. Their design is resilient to certain types of noise and does not require full fault-tolerance, making them experimentally viable [12] [27]. Within combinatorial chemistry, VQAs are primarily applied to problems such as calculating ground and excited states of molecules, simulating chemical dynamics, and optimizing molecular structures—tasks that are central to drug discovery and materials science [12] [25].
This technical guide explores the prospective pathway for these algorithms to achieve a quantum advantage in combinatorial chemistry. It provides a detailed examination of the core principles, current experimental protocols, and the evolving hardware and software landscape that will determine their practical impact.
Fundamental Principles of Variational Quantum Algorithms
The Variational Principle as a Foundation
The theoretical underpinning of VQAs is the variational principle of quantum mechanics. This principle states that for a given system Hamiltonian (H), the expectation value of energy for any trial quantum state (|ψ(θ)⟩) is always greater than or equal to the true ground state energy (E₀) [12]:
<ψ(θ)|H|ψ(θ)> ≥ E₀
This inequality provides a powerful method for approximating the ground state. By preparing a parameterized trial state, known as an ansatz, on a quantum computer and measuring its energy expectation value, one can use a classical optimizer to variationally minimize this energy. The minimized value provides an upper bound to the true ground state energy, and the corresponding state approximates the ground state wavefunction [12].
Algorithmic Workflow of a VQA
The execution of a VQA follows a well-defined hybrid quantum-classical loop, illustrated in the following workflow and described in detail below:
Figure 1. The hybrid quantum-classical workflow of a Variational Quantum Algorithm.
- Problem Initialization: The quantum computer is initialized in a default state, typically |00...0⟩. A parameterized quantum circuit (the ansatz), denoted by U(θ), is constructed to prepare the trial state |ψ(θ)⟩ = U(θ)|00...0⟩ [12].
- Quantum Execution and Measurement: The ansatz is executed on the quantum processor. The quantum state is then measured to estimate the expectation values of the Hamiltonian's components. This step provides the data needed to compute the total energy or other cost functions.
- Classical Optimization: The measured cost function, C(θ), is fed to a classical optimizer. The optimizer determines a new set of parameters θ to reduce the cost in the next iteration [12].
- Convergence Check: The loop continues until the cost function converges to a minimum within a predefined tolerance, indicating that an approximate solution to the problem has been found.
Core Algorithms and Methodologies
Key Algorithms for Chemistry
Two VQAs are particularly prominent in quantum computational chemistry:
- Variational Quantum Eigensolver (VQE): Primarily used to find the ground state (and sometimes excited states) energy of molecular systems. It has been experimentally demonstrated for small molecules like hydrogen, lithium hydride, and beryllium hydride [27] [25]. Its resilience to noise makes it a leading algorithm for the NISQ era.
- Quantum Approximate Optimization Algorithm (QAOA): While often applied to combinatorial optimization problems like Max-Cut, QAOA can also be tailored for chemistry problems framed as optimization tasks, such as certain types of molecular structure optimization [27].
The Ansatz in Quantum Chemistry
The ansatz is a critical choice that defines the expressibility of the quantum circuit and the efficiency of the optimization. It is an "educated guess" of the form of the quantum state [12]. In chemistry, common ansatz types include:
- Physically-Inspired Ansätze: Such as the Unitary Coupled Cluster (UCC) ansatz, which is adapted from a successful classical quantum chemistry method. It is highly accurate but can lead to deep quantum circuits.
- Hardware-Efficient Ansätze: Designed to match the native gate set and connectivity of a specific quantum processor. They generate shallow circuits but may be more prone to representing physically irrelevant states or suffering from optimization challenges like Barren Plateaus.
The ansatz allows the algorithm to restrict the search for the solution to a relevant subspace of the entire Hilbert space, making the problem tractable for the quantum computer [12].
Quantitative Landscape of Quantum Advantage
Algorithmic Resource Requirements
The table below summarizes the resource requirements and current status of key quantum algorithms relevant to combinatorial chemistry.
Table 1. Resource Requirements for Quantum Chemistry Algorithms.
| Algorithm | Primary Use Case | Qubit Count (Current Demo) | Target Industrial Qubit Needs | Key Challenge |
|---|---|---|---|---|
| VQE [25] [27] | Molecular Ground State Energy | ~10-20 qubits | ~2.7 million (for FeMoco) [25] | Ansatz depth, noise resilience |
| QAOA [27] [93] | Combinatorial Optimization | ~10s of qubits | Problem-dependent | Parameter optimization, convergence |
| Quantum Phase Estimation (QPE) [27] | Precision Energy & Dynamics | Requires fault-tolerance | Millions (logical qubits) | Deep circuits, full error-correction |
Hardware Error Correction Progress
Progress in quantum hardware is measured by the quality and quantity of qubits. Recent breakthroughs in error correction are crucial for future applications.
Table 2. 2025 Hardware and Error Correction Milestones [32].
| Company/Institution | Breakthrough | Reported Performance |
|---|---|---|
| Willow Chip (105 superconducting qubits) | Demonstrated exponential error reduction; completed a benchmark in 5 mins that would take a classical computer 10^25 years. | |
| IBM | Fault-Tolerant Roadmap | Quantum Starling system (200 logical qubits) targeted for 2029; plans for 100M error-corrected operations. |
| Microsoft | Majorana 1 (Topological) & Geometric Codes | 28 logical qubits encoded on 112 atoms; 1,000-fold error rate reduction. |
| Industry Wide | Algorithmic Fault Tolerance | Error rates pushed to 0.000015% per operation; error correction overhead reduced by up to 100x. |
Experimental Protocols and Research Toolkit
Detailed Protocol: VQE for Molecular Ground State Energy
This protocol outlines the steps for running a Variational Quantum Eigensolver experiment to compute the ground state energy of a molecule, a foundational task in combinatorial chemistry.
Step 1: Problem Formulation (Classical)
- Define the Molecule: Specify the molecular geometry (atomic species and positions) of the target molecule, e.g., a lithium hydride (LiH) molecule.
- Generate the Hamiltonian: Using a classical computer, perform a Hartree-Fock calculation on the defined molecule. Then, map the electronic Hamiltonian from a fermionic to a qubit representation using a transformation such as Jordan-Wigner or Bravyi-Kitaev. The result is a qubit Hamiltonian (H) expressed as a sum of Pauli strings: ( H = \sumi ci Pi ), where ( Pi ) are tensor products of Pauli operators (I, X, Y, Z).
Step 2: Ansatz Selection and Circuit Preparation (Classical)
- Choose an Ansatz: Select a parameterized quantum circuit U(θ). For chemical accuracy, a unitary coupled cluster with singles and doubles (UCCSD) ansatz is a common choice, though hardware-efficient ansätze may be used for shorter-depth circuits on NISQ devices.
- Initialize Parameters: Choose initial parameters (θ₀) for the ansatz. These can be random, zeros, or informed by a classical approximation (e.g., MP2 amplitudes).
Step 3: Quantum-Classical Loop
- State Preparation and Measurement: For the current set of parameters θ, execute the quantum circuit U(θ) on the quantum processor. Measure the expectation value ( \langle Pi \rangle ) for each Pauli term ( Pi ) in the Hamiltonian. This often requires repeating the circuit execution multiple times (shots) for each term to gather sufficient statistics.
- Compute Total Energy (Classical): Classically combine the measured expectation values with the coefficients ( ci ) to compute the total energy expectation value: ( E(θ) = \sumi ci \langle Pi \rangle ).
- Optimize Parameters (Classical): Feed E(θ) to a classical optimizer (e.g., SPSA, COBYLA, or gradient-based methods). The optimizer proposes a new set of parameters θ_new aimed at lowering the energy.
- Iterate: Steps 3a-3c are repeated until the energy E(θ) converges to a minimum, or a maximum number of iterations is reached.
The Scientist's Toolkit for Quantum Chemistry Experiments
Table 3. Essential "Research Reagent Solutions" for Quantum Computational Chemistry.
| Item / Concept | Function / Purpose | Example in Practice |
|---|---|---|
| Parameterized Quantum Circuit (Ansatz) [12] | Encodes the trial wavefunction; the function whose parameters are variationally optimized. | Unitary Coupled Cluster (UCC) ansatz for molecular ground states. |
| Classical Optimizer [12] | Finds parameters that minimize the cost function (e.g., energy). | Gradient-free optimizers like SPSA are robust to quantum hardware noise. |
| Qubit Hamiltonian [25] | The representation of the molecule's energy operator in the language of qubits. | A sum of Pauli terms generated via a Bravyi-Kitaev transformation of the electronic structure Hamiltonian. |
| Error Mitigation Techniques [93] | Post-processing methods to reduce the impact of noise without full error correction. | Zero-Noise Extrapolation (ZNE) or probabilistic error cancellation. |
| Conditional Value at Risk (CVaR) [93] | A loss function that focuses on the best outcomes in a distribution, improving optimization. | Using only the top 25% of measurement results to compute the energy expectation value. |
Pathways to Quantum Advantage
Technical and Resource Hurdles
The path to a demonstrable quantum advantage in combinatorial chemistry is contingent on overcoming significant hurdles:
- Qubit Scalability and Quality: While algorithms like VQE are designed for NISQ devices, industrial-scale problems require a massive number of high-quality qubits. For example, simulating the iron-molybdenum cofactor (FeMoco), crucial for nitrogen fixation, was recently estimated to require nearly 100,000 physical qubits with advanced error correction—a figure that remains far beyond current capabilities but is now considered a target rather than a fantasy [25] [32].
- Algorithmic Efficiency: The depth of quantum circuits and the trainability of VQAs remain central challenges. Issues such as Barren Plateaus, where the cost function gradient vanishes exponentially with system size, can halt optimization. Developing more efficient ansätze and optimization techniques is an active area of research [12].
- The Classical Counterpoint: The pursuit of quantum advantage exists in a dynamic landscape where classical algorithms also improve. The development of "quantum-inspired" algorithms—classical methods that run on high-performance computers but mimic quantum approaches—poses a competitive benchmark that quantum algorithms must surpass to prove their value [25].
Application Frontiers in Combinatorial Chemistry
The following diagram maps the pathway from current experimental capabilities to future applications requiring a full quantum advantage, highlighting the key transitional milestones.
Figure 2. The projected pathway to quantum advantage in combinatorial chemistry, from current small-scale demonstrations to the simulation of industrially relevant molecules.
Potential application frontiers that could define quantum advantage include:
- Drug Discovery: Precisely simulating protein-ligand interactions and the dynamics of large biomolecules, such as the KRAS protein implicated in cancers, which has seen early-stage quantum simulation [25].
- Materials Design: Modeling complex materials with strong electron correlations, such as high-temperature superconductors or novel battery electrolytes, which are notoriously difficult for classical methods like Density Functional Theory (DFT) [32] [25].
- Reaction Optimization: Using quantum algorithms to explore vast combinatorial reaction spaces to identify optimal pathways and conditions for synthetic chemistry, potentially revolutionizing the development of industrial catalysts [25].
The prospects for achieving a quantum advantage in combinatorial chemistry are intrinsically linked to the co-evolution of algorithmic innovation and hardware progress. Variational Quantum Algorithms provide a tangible, near-term framework for exploring these prospects on existing hardware. The community has moved beyond proof-of-concept demonstrations for diatomic molecules and is now tackling the steep path toward simulating industrially significant chemical systems.
The transition from laboratory curiosity to commercial technology will be determined by overcoming the dual challenges of qubit scalability and algorithmic efficiency. As error correction milestones continue to be achieved and new algorithmic strategies like CVaR and better ansätze are developed, the timeline for practical quantum advantage in combinatorial chemistry is expected to solidify, potentially within the next decade for specific, high-impact problems.
Variational Quantum Algorithms (VQAs) have emerged as the leading paradigm for harnessing the computational potential of Noisy Intermediate-Scale Quantum (NISQ) devices. These hybrid quantum-classical algorithms are particularly promising for applications in combinatorial chemistry research, where they aim to solve electronic structure problems that are computationally intractable for classical computers. The core functionality of VQAs involves using a quantum processor to prepare and measure parameterized quantum states, while a classical optimizer adjusts parameters to minimize a cost function, typically the energy expectation value of a molecular Hamiltonian. For drug development professionals and computational chemists, VQAs offer a pathway to more accurate simulation of molecular systems, reaction mechanisms, and catalytic processes, potentially accelerating the discovery of novel pharmaceuticals and materials.
Despite their promise, VQAs face significant challenges that must be addressed before they can deliver practical quantum advantage in real-world chemistry applications. Three interconnected areas represent the critical frontiers for development: enhancing algorithmic scalability to handle chemically relevant system sizes, implementing effective error mitigation strategies to compensate for NISQ device imperfections, and refining hybrid algorithm architectures to optimally leverage classical and quantum resources. This technical guide examines recent advances and future directions across these domains, providing researchers with the methodologies and frameworks needed to advance computational chemistry research using quantum algorithms.
Scalability
The scalability of VQAs refers to their ability to handle problems of increasing size and complexity, particularly those relevant to pharmaceutical research where molecules of interest often require substantial quantum resources for accurate simulation. Current NISQ devices typically offer 50-500 qubits, which limits the size of molecular systems that can be directly simulated without algorithmic innovations.
Circuit Parallelization Framework
A promising approach for overcoming qubit limitations involves circuit slicing, where a large quantum circuit representing a target molecular system is divided into smaller, independently executable subcircuits called slices. The formal procedure can be described as follows:
Consider a parameterized quantum circuit of a VQA defined on N qubits, with a quantum register of only n available qubits (where n < N). The slicing methodology involves:
- Problem Decomposition: Inspecting the target molecular Hamiltonian to identify r computationally separable subproblems that can be implemented as smaller parameterized quantum circuits.
- Slice Implementation: Each slice must be executable on at most n qubits, with the sum of qubits across all slices equaling N.
- Global Optimization: The classical optimization routine simultaneously coordinates all slices, optimizing a global objective function that approximates the original problem [6].
This method transforms the optimization of a single black-box function on a 2^N-dimensional space into the parallel optimization of r black-box functions on smaller spaces, dramatically reducing quantum hardware requirements while preserving the essential structure of the computational chemistry problem.
Comparative Analysis of Scalability Approaches
Table 1: Approaches for Enhancing VQA Scalability in Chemical Computation
| Approach | Key Methodology | Advantages | Chemical Applications |
|---|---|---|---|
| Circuit Parallelization | Splitting large circuits into smaller, parallel-executable slices [6] | Enables solving problems larger than available qubits; reduces circuit depth | Large molecular systems; protein-ligand binding simulations |
| Contextual Subspaces | Hybrid quantum-classical partitioning of molecular Hamiltonians [6] | Significantly reduces required qubits while maintaining chemical accuracy | Molecular ground state energy calculations |
| Tensor Network Pretraining | Using classical tensor networks to initialize VQA parameters [94] | Faster convergence; reduced quantum resource requirements | Strongly correlated molecular systems |
Quantum Resource Requirements
Table 2: Quantum Resource Requirements for Molecular Simulations
| Molecule | Qubits (Full CI) | Qubits (Parallelized) | Circuit Depth Reduction |
|---|---|---|---|
| H₂O | 14 | 2 × 7 | 35% [6] |
| N₂ | 20 | 4 × 5 | 42% [6] |
| F₂ | 20 | 4 × 5 | 40% [6] |
| Pharmaceutical Target | ~100 | 10 × 10 | ~50% (estimated) |
Figure 1: Parallelization framework for scalable molecular simulation, showing how a large target molecule is decomposed into smaller slices for parallel quantum processing, with a classical optimizer coordinating through a global objective function.
Error Mitigation
Quantum error mitigation (QEM) comprises a suite of techniques designed to improve the accuracy of quantum computations without the qubit overhead required by full quantum error correction. In computational chemistry applications, where precise energy calculations are essential for predicting reaction pathways and molecular properties, effective error mitigation is particularly critical.
Multireference State Error Mitigation
The Multireference State Error Mitigation (MREM) method represents a significant advancement for handling strongly correlated molecular systems, where traditional single-reference approaches fail. The protocol extends the original Reference-state Error Mitigation (REM) method, which uses a single Hartree-Fock state as a reference, by incorporating multiple Slater determinants to better approximate strongly correlated ground states [95].
Experimental Protocol for MREM Implementation:
Reference State Selection:
- Perform classical multiconfigurational calculations (e.g., CASSCF, DMRG) to identify dominant Slater determinants in the target molecular wavefunction.
- Select a truncated set of determinants that collectively provide sufficient overlap with the true ground state, typically focusing on those with the largest coefficients.
Quantum Circuit Preparation:
- Implement the multireference state using Givens rotation circuits, which systematically construct linear combinations of Slater determinants while preserving physical symmetries (particle number, spin projection) [95].
- For a reference state comprising K Slater determinants, the circuit requires at most K Givens rotation networks, with each network preparing one determinant from the initial Hartree-Fock state.
Error Mitigation Execution:
- Prepare and measure both the target state (|ψ(θ)⟩) and multireference states (|ϕ₁⟩, |ϕ₂⟩, ..., |ϕₖ⟩) on the quantum device.
- For each state, collect measurement statistics to estimate the energy expectation values Eₜₐᵣₜₑₜ and Eₘᵣ.
- Classically compute the exact energies for the multireference states (Eₘᵣ₍ₑₓₐ𝒸ₜ₎) using conventional methods.
Error Correction Application:
- Apply the MREM correction to obtain the mitigated energy: Eₘᵢₜᵢ₉ₐₜₑ𝒹 = Eₜₐᵣₜₑₜ + Σᵢ wᵢ (Eᵢ₍ₑₓₐ𝒸ₜ₎ - Eᵢ₍ₙₒᵢₛᵧ₎), where wᵢ are weights based on the importance of each reference state [95].
Error Mitigation Performance
Table 3: Performance Comparison of Error Mitigation Techniques for Molecular Energy Calculations
| Method | Theoretical Basis | Sampling Overhead | Accuracy Improvement | Limitations |
|---|---|---|---|---|
| REM | Single-reference (Hartree-Fock) error calibration [95] | Low | ~60% for weakly correlated systems [95] | Fails for strong correlation |
| MREM | Multireference error calibration [95] | Moderate | ~85% for strongly correlated systems [95] | Requires classical multireference calculation |
| Zero-Noise Extrapolation | Extrapolating to zero error rate from multiple noise levels [95] | High | ~70% for medium-depth circuits | Exponential sampling cost |
| Probabilistic Error Cancellation | Applying inverse error operations in post-processing [95] | Very High | ~80% with complete noise characterization | Exponential sampling cost |
Table 4: Essential Computational Tools for Quantum Error Mitigation in Chemistry Research
| Tool/Resource | Function | Implementation Notes |
|---|---|---|
| Givens Rotation Circuits | Prepares multireference states from initial Hartree-Fock state [95] | Preserves particle number and spin symmetry; requires O(K×M²) gates for K determinants over M orbitals |
| Classical Multireference Solver | Identifies dominant Slater determinants for target molecular system | CASSCF or DMRG calculations provide reference states for MREM |
| Shot Budget Manager | Allocates measurement samples across mitigation techniques | Critical for managing exponential sampling overhead in some methods |
| Noise Characterization Kit | Profiles device-specific error rates for extrapolation methods | Includes randomized benchmarking and gate set tomography |
Figure 2: MREM workflow for molecular energy calculation, showing integration of classical multireference analysis with quantum measurement and error mitigation.
Hybrid Algorithms
Hybrid quantum-classical algorithms represent the dominant paradigm for NISQ-era quantum computation, strategically partitioning computational workloads between quantum and classical processors to maximize their respective strengths. For combinatorial chemistry research, these algorithms enable the solution of electronic structure problems that exceed the capabilities of purely classical computational methods.
Advanced Hybrid Architectures
Recent research has produced several innovative hybrid architectures that extend beyond standard VQE approaches:
Neural-Guided VQAs: The sVQNHE framework introduces a neural-guided variational quantum algorithm that decouples amplitude and sign learning across classical and quantum modules. This approach employs shallow quantum circuits composed of commuting diagonal gates to model quantum phase information, while a classical neural network learns the amplitude distribution and guides circuit optimization in a bidirectional feedback loop. For the 6-qubit J₁-J₂ model, this method reduces the mean absolute error by 98.9% and suppresses variance by 99.6% relative to a baseline neural network, while requiring nearly 19× fewer optimization steps than a standard hardware-efficient VQE [96].
Variational Quantum Imaginary Time Evolution: VarQITE applies the mathematical formalism of imaginary time evolution to ground-state preparation, making it particularly suitable for constrained combinatorial optimization problems. When applied to the Multiple Knapsack Problem, VarQITE achieves significantly lower mean optimality gaps compared to QAOA and other conventional methods. Hamiltonian coefficient scaling can further reduce optimization costs and accelerate convergence [5].
Quantum Annealing Variants: Beyond gate-based approaches, variants of quantum annealing have shown promise for specific classes of chemical optimization problems. By using the spin coherent-state path integral to shape the geometry of quantum adiabatic evolution, these methods provide orders-of-magnitude improvements in the probability to measure optimal solutions, relative to linear protocols, particularly for hard problem instances with first-order quantum phase transitions [97].
Experimental Protocol: Neural-Guided VQA Implementation
For researchers seeking to implement the neural-guided approach for molecular electronic structure calculations:
Circuit Initialization:
- Design a shallow quantum circuit with commuting diagonal gates focused on capturing sign structure information.
- Initialize a classical neural network (typically a feedforward or recurrent architecture) to model the amplitude distribution of the target wavefunction.
Hybrid Optimization Loop:
- Quantum Phase Estimation: Run the parameterized quantum circuit to estimate phase information for the current molecular Hamiltonian.
- Classical Amplitude Learning: Update the neural network parameters to minimize the energy expectation value using the quantum phase information.
- Bidirectional Feedback: Use the neural network's output to guide quantum circuit parameter updates, creating a closed-loop optimization system.
Convergence Monitoring:
- Track both the energy expectation value and its variance across optimization steps.
- Implement early stopping if variance plateaus or begins increasing, indicating potential overfitting or convergence issues.
This architecture has demonstrated particular effectiveness for molecular systems with severe sign problems that challenge Monte Carlo-based methods, achieving significantly improved resource efficiency compared to standard VQE approaches [96].
Hybrid Algorithm Performance Metrics
Table 5: Performance Comparison of Hybrid Algorithms for Chemical and Optimization Problems
| Algorithm | Application Domain | Key Metric | Performance Improvement |
|---|---|---|---|
| Standard VQE | Molecular ground state energy [7] | Energy accuracy | Baseline for comparison |
| Neural-Guided sVQNHE | Many-body systems with sign problem [96] | Mean absolute error | 98.9% reduction vs. baseline [96] |
| VarQITE | Constrained combinatorial optimization [5] | Optimality gap | Significant reduction vs. QAOA [5] |
| Enhanced Annealing | Hard optimization instances [97] | Probability of optimal solution | Orders-of-magnitude improvement [97] |
Figure 3: Neural-guided hybrid algorithm architecture showing bidirectional feedback between classical neural network and quantum circuit for molecular energy calculation.
The future of variational quantum algorithms in combinatorial chemistry research hinges on simultaneous advances across scalability, error mitigation, and hybrid algorithmic architectures. The research community has developed promising pathways forward in each domain: circuit parallelization methods that transcend immediate qubit limitations, multireference error mitigation techniques that address the critical challenge of strong electron correlation, and increasingly sophisticated hybrid architectures that optimally leverage both classical and quantum computational resources.
For drug development professionals and computational chemists, these advances collectively enable the simulation of increasingly complex molecular systems with higher accuracy and efficiency. While practical quantum advantage for pharmaceutical applications remains on the horizon, the methodological improvements documented in this guide represent essential stepping stones toward that goal. Future research will likely focus on tighter integration of these approaches—developing scalable error-mitigated hybrid algorithms that can efficiently tackle the challenging electronic structure problems prevalent in drug discovery and materials design. As quantum hardware continues to evolve in scale and fidelity, the algorithmic foundations being established today will position researchers to fully exploit these technological advances for transformative advances in combinatorial chemistry.
Conclusion
Variational Quantum Algorithms represent a paradigm shift for combinatorial chemistry, offering a viable path to solving problems that are intractable for classical computers. While challenges such as noise and training difficulties persist, VQAs have already demonstrated concrete utility in real-world drug discovery applications, from simulating prodrug activation to modeling covalent inhibitor interactions. The integration of VQAs into hybrid quantum-classical pipelines marks a significant step toward practical quantum-enhanced drug design. Future progress hinges on developing more robust error mitigation strategies, creating chemically-inspired ansätze, and scaling these methods to larger, commercially-relevant molecular systems. As quantum hardware continues to advance, VQAs are poised to move from specialized tools to central components in the computational chemist's arsenal, potentially revolutionizing the pace and precision of biomedical research and clinical development.
References
- 1. Noisy intermediate-scale quantum computing [https://en.wikipedia.org/wiki/Noisy_intermediate-scale_quantum_computing]
- 2. Variational Quantum Algorithms: From Theory to NISQ-Era ... [https://www.preprints.org/manuscript/202508.1482]
- 3. Quantum Computing in the NISQ era and beyond [https://quantum-journal.org/papers/q-2018-08-06-79/]
- 4. Quantum Computing for Chemical Applications: Variational ... [https://link.springer.com/article/10.1007/s41745-025-00488-2]
- 5. Solving Constrained Combinatorial Optimization Problems ... [https://arxiv.org/abs/2504.12607]
- 6. Parallel circuit implementation of variational quantum ... [https://www.nature.com/articles/s41534-025-00982-6]
- 7. Variational Quantum Algorithms: The Practical Beginner's ... [https://medium.com/@raktims2210/variational-quantum-algorithms-the-practical-beginners-guide-to-nisq-quantum-computing-for-ai-81b27500276c]
- 8. A benchmarking study of quantum algorithms for ... [https://www.nature.com/articles/s41534-024-00856-3]
- 9. Exact gradient for universal cost functions in variational ... [https://arxiv.org/html/2509.17822v1]
- 10. What is Variational Quantum Algorithm [https://www.quera.com/glossary/variational-quantum-algorithm]
- 11. Variational quantum algorithm [https://www.quandela.com/resources/quantum-computing-glossary/variational-quantum-algorithm/]
- 12. Variational Quantum Algorithms [https://medium.com/@qcgiitr/variational-quantum-algorithms-66367053a2f3]
- 13. Variational Quantum Machine Learning Algorithms [https://www.quantum-machine-learning.com/page/variational-qml-algorithms/]
- 14. Parameterized quantum circuits as universal generative ... [https://www.nature.com/articles/s41534-025-01064-3]
- 15. [2401.10121] A Novel Noise-Aware Classical Optimizer for ... [https://arxiv.org/abs/2401.10121]
- 16. Variational quantum eigensolver [https://en.wikipedia.org/wiki/Variational_quantum_eigensolver]
- 17. Variational Quantum Algorithms for Chemical Simulation ... [https://arxiv.org/abs/2211.07854]
- 18. Quantum Computing for Chemical Applications: Variational ... [https://chemrxiv.org/engage/chemrxiv/article-details/68b92f9123be8e43d6deb787]
- 19. A brief overview of VQE | PennyLane Demos [https://pennylane.ai/qml/demos/tutorial_vqe]
- 20. [2507.20532] Quantum Portfolio Optimization with Expert ... [https://arxiv.org/abs/2507.20532]
- 21. Extending QAOA-GPT to Higher-Order Quantum ... [https://arxiv.org/html/2511.07391v1]
- 22. Intro to QAOA | PennyLane Demos [https://pennylane.ai/qml/demos/tutorial_qaoa_intro]
- 23. Variational Quantum Algorithms for Combinatorial ... [https://arxiv.org/abs/2407.06421]
- 24. The Variational Quantum Eigensolver: a review of methods ... [https://arxiv.org/abs/2111.05176]
- 25. Will quantum computing be chemistry's next AI? - C&EN [https://cen.acs.org/business/quantum-computing-chemistrys-next-AI/103/web/2025/11]
- 26. Quantum machine learning of molecular energies with hybrid ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00222b]
- 27. Quantum Algorithms Guide: Principles, Types, and Use ... [https://www.spinquanta.com/news-detail/the-ultimate-guide-to-quantum-algorithms]
- 28. New Hybrid Quantum–Classical Computing Approach Used to ... [https://researchimpact.caltech.edu/research-news/new-hybrid-quantumclassical-computing-approach-used-to-study-chemical-systems]
- 29. Scaling for quantum advantage and beyond [https://www.ibm.com/quantum/blog/qdc-2025]
- 30. Quantum Computing Roadmaps & Predictions of Leading ... [https://thequantuminsider.com/2025/05/16/quantum-computing-roadmaps-a-look-at-the-maps-and-predictions-of-major-quantum-players/]
- 31. Our quantum hardware: the engine for verifiable ... [https://blog.google/technology/research/quantum-hardware-verifiable-advantage/]
- 32. Quantum Computing Industry Trends 2025: A Year of ... [https://www.spinquanta.com/news-detail/quantum-computing-industry-trends-2025-breakthrough-milestones-commercial-transition]
- 33. A verifiable quantum advantage [https://research.google/blog/a-verifiable-quantum-advantage/]
- 34. Mind the gaps: The fraught road to quantum advantage [https://arxiv.org/html/2510.19928v2]
- 35. A hybrid quantum computing pipeline for real world drug ... [https://www.nature.com/articles/s41598-024-67897-8]
- 36. Ground state energy and molecular properties calculations [https://www.sciencedirect.com/science/article/pii/S2352492823024510]
- 37. A hybrid quantum computing pipeline for real world drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11266395/]
- 38. Generative quantum combinatorial optimization by means of a ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00138b]
- 39. [2012.09265] Variational Quantum Algorithms [https://arxiv.org/abs/2012.09265]
- 40. A quantum for spin algorithm : a Bayesian exchange... chemistry [https://pmc.ncbi.nlm.nih.gov/articles/PMC8179312/]
- 41. Quantum intelligence in drug discovery: Advancing insights ... [https://www.sciencedirect.com/science/article/abs/pii/S135964462500176X]
- 42. Quantum computing in life sciences and drug discovery [https://www.mckinsey.com/industries/life-sciences/our-insights/the-quantum-revolution-in-pharma-faster-smarter-and-more-precise]
- 43. How quantum computing is changing drug development ... [https://www.weforum.org/stories/2025/01/quantum-computing-drug-development/]
- 44. Targeting KRAS mutations: orchestrating cancer evolution ... [https://www.nature.com/articles/s41392-025-02473-8]
- 45. Advances in biomarkers of resistance to KRAS mutation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12508421/]
- 46. Quantitative Systems Pharmacology Analysis of KRAS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5980551/]
- 47. A general framework for active space embedding methods ... [https://www.nature.com/articles/s41524-024-01477-2]
- 48. Quantum Information-Assisted Complete Active Space ... [https://arxiv.org/html/2309.01676v2]
- 49. Quantum computing makes waves in drug discovery [https://www.stjude.org/research/progress/2025/quantum-computing-makes-waves-in-drug-discovery.html]
- 50. Hybrid Quantum-Classical simulations (QM/MM) with CP2K ... [https://manual.gromacs.org/current/reference-manual/special/qmmm.html]
- 51. Machine Learning Enhanced Calculation of Quantum- ... [https://arxiv.org/html/2503.03955v1]
- 52. From Wafer‑Scale Protein Simulations to Hybrid Quantum ... [https://beit.tech/blog/from-wafer-scale-protein-simulations-to-hybrid-quantum-chemistry-beit-s-latest-breakthroughs]
- 53. Variational quantum algorithms (Chapter 20) [https://resolve.cambridge.org/core/books/quantum-algorithms/variational-quantum-algorithms/0F3154CFD9A96737B25D36D7E867BE5C]
- 54. Can Classical Initialization Help Variational Quantum Circuits Escape the Barren Plateau? The views expressed in this article are those of the authors and do not represent the views of Wells Fargo. This article is for informational purposes only. Nothing contained in this article should be construed as investment advice. Wells Fargo makes no express or implied warranties and expressly disclaims all legal, tax, and accounting implications related to this article. [https://arxiv.org/html/2508.18497]
- 55. A Survey of Methods for Mitigating Barren Plateaus for Parameterized Quantum Circuits [https://arxiv.org/html/2406.14285v1]
- 56. Engineered dissipation to mitigate barren plateaus [https://www.nature.com/articles/s41534-024-00875-0]
- 57. Investigating and Mitigating Barren Plateaus in Variational ... [https://arxiv.org/abs/2407.17706]
- 58. Fourier Analysis of Parameterized Quantum Circuits and the Barren Plateau Problem [https://arxiv.org/html/2309.06740]
- 59. Chemical Apps 2025 – Conference Tagline - ORNL Events [https://events.ornl.gov/chemicalapps2025/]
- 60. Quantum Variational Formula Explained with Examples [https://www.spinquanta.com/news-detail/quantum-variational-formula-explained-with-examples]
- 61. Variational quantum algorithms: fundamental concepts ... [https://link.springer.com/article/10.1007/s11128-024-04438-2]
- 62. Designing Minimalistic Variational Quantum Ansatz ... [https://arxiv.org/html/2501.16776v1]
- 63. A Depth-Independent Linear Chain Ansatz for Large-Scale ... [https://arxiv.org/html/2509.17296v1]
- 64. Solving Constrained Combinatorial Optimization Problems ... [https://arxiv.org/html/2504.12607v2]
- 65. Enhancing Hybrid Methods in Parameterized Quantum ... [https://arxiv.org/html/2510.08142v1]
- 66. Optimizing Parameters of Quantum Circuits with Sparsity ... [https://www.ijcai.org/proceedings/2025/680]
- 67. Enhancing Hybrid Methods in Parameterized Quantum ... [https://arxiv.org/abs/2510.08142]
- 68. Structure optimization for parameterized quantum circuits [https://quantum-journal.org/papers/q-2021-01-28-391/]
- 69. Princeton's new quantum chip built for scale [https://materials.princeton.edu/news/2025/princeton%E2%80%99s-new-quantum-chip-built-scale]
- 70. Quantum decoherence [https://en.wikipedia.org/wiki/Quantum_decoherence]
- 71. Quantum Decoherence: The Barrier to Quantum Computing [https://www.bluequbit.io/blog/quantum-decoherence]
- 72. Refining Noise Mitigation in NISQ Hardware Through Qubit ... [https://arxiv.org/html/2503.10204v1]
- 73. Quantum Error Mitigation in the NISQ Era [https://medium.com/@raktims2210/quantum-error-mitigation-in-the-nisq-era-87af568290e5]
- 74. An Overview of Noise-Adaptive Quantum Algorithms [https://thequantuminsider.com/2025/06/14/an-overview-of-noise-adaptive-quantum-algorithms/]
- 75. How does the electronic structure Hamiltonian scales with ... [https://chemistry.stackexchange.com/questions/159424/how-does-the-electronic-structure-hamiltonian-scales-with-basis-sets-and-the-num]
- 76. Comparing performance of variational quantum algorithm ... [https://arxiv.org/abs/2507.17614]
- 77. Accelerated variational algorithms for digital ... [https://quantum-journal.org/papers/q-2020-09-16-324/]
- 78. Hamiltonian Computational Chemistry: Geometrical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11120360/]
- 79. Accelerating variational quantum algorithms with multiple ... [https://arxiv.org/abs/2106.12819]
- 80. Variational quantum algorithm for enhanced continuous ... [https://www.nature.com/articles/s41534-024-00947-1]
- 81. Hartree–Fock method [https://en.wikipedia.org/wiki/Hartree%E2%80%93Fock_method]
- 82. [For Beginners] What is Density Functional Theory (DFT)? [https://matlantis.com/en/resources/blog/dft/]
- 83. Gold-Standard Chemical Database 138 (GSCDB138) [https://arxiv.org/html/2508.13468v1]
- 84. CASSCF Calculation - an overview [https://www.sciencedirect.com/topics/chemistry/casscf-calculation]
- 85. Towards Compact Wavefunctions from Quantum-Selected ... [https://arxiv.org/html/2509.02525v2]
- 86. Benchmarking a foundation potential against quantum ... [https://ui.adsabs.harvard.edu/abs/2025arXiv251024063C/abstract]
- 87. A Comparative Study of Quantum Optimization Techniques ... [https://ui.adsabs.harvard.edu/abs/2025arXiv250312121S/abstract]
- 88. Benchmarking Variational Quantum Algorithms for ... [https://arxiv.org/abs/2408.03073]
- 89. Quality Diversity for Variational Quantum Circuit Optimization [https://ojs.aaai.org/index.php/ICAPS/article/view/36139]
- 90. Algorithms used in Quantum Optimization [https://github.com/SMU-Quantum/quantum-optimization-algorithms]
- 91. Workshops Abstracts - IEEE Quantum Week [https://qce.quantum.ieee.org/2025/workshops-abstracts/]
- 92. Comparing performance of variational quantum algorithm ... [https://arxiv.org/html/2507.17614v1]
- 93. Searching for quantum advantage in mathematical ... [https://www.ibm.com/quantum/blog/optimization-white-paper]
- 94. [PDF] An introduction to variational quantum algorithms for ... [https://www.semanticscholar.org/paper/An-introduction-to-variational-quantum-algorithms-Grange-Poss/4f101d9e03ff2b1abb5f8c5f0ef70358403f5ed6]
- 95. Multireference error mitigation for quantum computation of ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00202h]
- 96. A Neural-Guided Variational Quantum Algorithm for ... [https://arxiv.org/abs/2507.07555]
- 97. Quantum combinatorial optimization beyond the variational ... [https://ui.adsabs.harvard.edu/abs/2025PhRvA.111c2411B/abstract]